Генетическое расстройство - Genetic disorder
| Генетическое расстройство | |
|---|---|
 | |
| Мальчик с Синдром Дауна, одно из самых распространенных генетических заболеваний | |
| Специальность | Медицинская генетика |
А генетическое расстройство проблема со здоровьем, вызванная одной или несколькими аномалиями в геном. Это может быть вызвано мутация в единственном ген (моногенные) или множественные гены (полигенные) или хромосомная аномалия. Хотя полигенные расстройства являются наиболее распространенными, этот термин в основном используется при обсуждении расстройств, имеющих единственную генетическую причину, либо в гене, либо в хромосома.[1][2] Ответственная мутация может произойти спонтанно до эмбриональное развитие (а de novo мутация), или это может быть унаследованный от двух родителей, которые являются носителями дефектного гена (аутосомно-рецессивный по наследству) или от родителя с заболеванием (аутосомно-доминантный наследование). Некоторые расстройства вызваны мутацией Х хромосома и имеют Х-связанный наследование. Очень немногие расстройства передаются по наследству Y-хромосома или же митохондриальная ДНК.[3]
Известно более 6000 генетических заболеваний,[4] и новые генетические нарушения постоянно описываются в медицинской литературе.[5] Примерно 1 из 50 человек страдает известным моногенным заболеванием, в то время как примерно 1 из 263 человек страдает от хромосомное заболевание.[6] Около 65% людей имеют проблемы со здоровьем в результате врожденных генетических мутаций.[6] Из-за значительного количества генетических нарушений примерно каждый 21 человек страдает генетическим заболеванием, классифицируемым как "редкий "(обычно определяется как поражение менее 1 из 2 000 человек). Большинство генетических нарушений редки сами по себе.[5][7]
Рак вызваны генетическими мутациями, но обычно не упоминаются при упоминании генетических нарушений, поскольку большинство из них не являются наследственными (хотя предрасположенности и онкологические синдромы существовать).[8]
Один ген
| Распространенность заболевания (приблизительная) | |
|---|---|
| Аутосомно-доминантный | |
| Семейная гиперхолестеринемия | 1 из 500[9] |
| Поликистоз почек | 1 из 750[10] |
| Нейрофиброматоз I типа | 1 из 2500[11] |
| Наследственный сфероцитоз | 1 из 5000 |
| Синдром Марфана | 1 из 4000[12] |
| болезнь Хантингтона | 1 из 15 000[13] |
| Аутосомно-рецессивный | |
| Серповидно-клеточная анемия | 1 из 625[14] |
| Кистозный фиброз | 1 из 2000 |
| Болезнь Тея – Сакса | 1 из 3000 |
| Фенилкетонурия | 1 из 12 000 |
| Мукополисахаридозы | 1 из 25 000 |
| Дефицит липазы лизосомальной кислоты | 1 из 40 000 |
| Болезни накопления гликогена | 1 из 50 000 |
| Галактоземия | 1 из 57000 |
| Х-связанный | |
| Мышечная дистрофия Дюшенна | 1 из 5000 |
| Гемофилия | 1 из 10 000 |
| Значения указаны для живорожденных. | |
А моногенное заболевание (или же моногенное расстройство) является результатом одного мутировавший ген. Одногенные нарушения могут передаваться последующим поколениям несколькими способами. Геномный импринтинг и однопородная дисомия Однако это может повлиять на шаблоны наследования. Разделение между рецессивный и доминантный типы не являются "жесткими и быстрыми", хотя различия между аутосомный и Х-связанный типы есть (поскольку последние типы различаются исключительно на основе хромосомного расположения гена). Например, обычная форма карликовость, ахондроплазия, обычно считается доминантным заболеванием, но у детей с двумя генами ахондроплазии наблюдается тяжелое и обычно летальное заболевание скелета, носителями которого можно считать ахондроплазию. Серповидноклеточная анемия также считается рецессивным состоянием, но гетерозиготный перевозчики имеют повышенную устойчивость к малярия в раннем детстве, что можно охарактеризовать как родственное доминирующее состояние.[15] Если пара, в которой один или оба партнера являются больными или носителями моногенного заболевания, желает иметь ребенка, они могут сделать это через in vitro оплодотворение, которое позволяет провести преимплантационную генетическую диагностику, чтобы проверить, есть ли у эмбриона генетическое заболевание.[16]
Самый врожденный метаболический расстройства, известные как врожденные нарушения обмена веществ в результате дефектов одного гена. Многие такие единичные генные дефекты могут снизить физическую форму пораженных людей и, следовательно, присутствуют в популяции с меньшей частотой по сравнению с тем, что можно было бы ожидать на основе простых вероятностных расчетов.[17]
Аутосомно-доминантный
Для человека, страдающего аутосомно-доминантным заболеванием, потребуется только одна мутированная копия гена. У каждого пострадавшего обычно есть один пострадавший родитель.[18]:57 Вероятность того, что ребенок унаследует мутировавший ген, составляет 50%. Аутосомно-доминантные состояния иногда уменьшаются пенетрантность Это означает, что хотя требуется только одна мутированная копия, не все люди, унаследовавшие эту мутацию, развивают болезнь. Примеры этого типа расстройства: болезнь Хантингтона,[18]:58 нейрофиброматоз 1 типа, нейрофиброматоз 2 типа, Синдром Марфана, наследственный неполипозный колоректальный рак, наследственные множественные экзостозы (аутосомно-доминантное заболевание с высокой проникающей способностью), туберозный склероз, Болезнь фон Виллебранда, и острая перемежающаяся порфирия. Врожденные дефекты также называют врожденными аномалиями.
Аутосомно-рецессивный
Две копии гена должны быть мутированы, чтобы человек пострадал от аутосомно-рецессивного заболевания. У пострадавшего человека обычно есть здоровые родители, каждый из которых несет одну копию мутировавшего гена и упоминается как генетические носители. Каждый родитель с дефектным геном обычно не имеет симптомов.[19] Два здоровых человека, каждый из которых несет по одной копии мутировавшего гена, имеют 25% -ный риск при каждой беременности иметь ребенка, пораженного этим заболеванием. Примеры этого типа расстройства: альбинизм, дефицит ацил-КоА-дегидрогеназы со средней длиной цепи, кистозный фиброз, серповидноклеточная анемия, Болезнь Тея – Сакса, Болезнь Ниманна – Пика, спинальная мышечная атрофия, и Синдром Робертса. Некоторые другие фенотипы, например влажный или сухой ушная сера, также определяются по аутосомно-рецессивному типу.[20][21] Некоторые аутосомно-рецессивные расстройства встречаются часто, потому что в прошлом перенос одного из дефектных генов приводил к небольшая защита против инфекционного заболевания или токсин Такие как туберкулез или же малярия.[22] Такие расстройства включают кистозный фиброз,[23] серповидноклеточная анемия,[24] фенилкетонурия[25] и талассемия.[26]

Х-сцепленный доминантный
Х-сцепленные доминантные расстройства вызваны мутациями в генах Х хромосома. Только несколько заболеваний имеют этот образец наследования, ярким примером которого является Х-сцепленный гипофосфатемический рахит. Этим расстройствам подвержены как мужчины, так и женщины, причем мужчины обычно страдают более серьезно, чем женщины. Некоторые Х-сцепленные доминантные состояния, такие как Синдром Ретта, недержание мочи тип 2 и Синдром Айкарди, обычно смертельны для мужчин в утробе или вскоре после рождения, и поэтому преимущественно наблюдаются у женщин. Исключение составляют исключительно редкие случаи, когда мальчики с Синдром Клайнфельтера (44 + xxy) также наследуют Х-сцепленное доминантное состояние и проявляют симптомы, более похожие на женские по тяжести заболевания. Вероятность передачи Х-сцепленного доминантного расстройства у мужчин и женщин различается. Все сыновья мужчины с Х-сцепленным доминантным расстройством не пострадают (поскольку они получают Y-хромосому своего отца), но все его дочери унаследуют это заболевание. Женщина с Х-сцепленным доминантным расстройством имеет 50% -ный шанс иметь пораженный плод при каждой беременности, хотя в таких случаях, как пигментное недержание мочи, обычно жизнеспособны только потомки женского пола.
Х-сцепленный рецессивный
Х-сцепленные рецессивные состояния также вызваны мутациями в генах Х-хромосомы. Мужчины страдают намного чаще, чем женщины, потому что у них есть только одна Х-хромосома, необходимая для этого состояния. Вероятность передачи заболевания у мужчин и женщин разная. Сыновья мужчины с Х-сцепленным рецессивным заболеванием не пострадают (поскольку они получают Y-хромосому своего отца), но его дочери будут носителями одной копии мутировавшего гена. Женщина, которая является носительницей Х-сцепленного рецессивного расстройства (XрИкср) имеет 50% шанс иметь сыновей, которые затронуты, и 50% вероятность иметь дочерей, которые являются носителями одной копии мутировавшего гена. Х-сцепленные рецессивные состояния включают серьезные заболевания гемофилия А, Мышечная дистрофия Дюшенна, и Синдром Леша – Найхана, а также общие и менее серьезные состояния, такие как облысение по мужскому типу и красно-зеленый дальтонизм. Х-сцепленные рецессивные состояния могут иногда проявляться у женщин из-за: искаженная X-инактивация или моносомия X (Синдром Тернера ).
Y-связанный
Y-сцепленные расстройства вызваны мутациями в Y-хромосоме. Эти состояния могут передаваться только от гетерогаметного пола (например, люди мужского пола) потомству того же пола. Проще говоря, это означает, что Y-сцепленные расстройства у людей могут передаваться только от мужчин их сыновьям; самки никогда не могут быть затронуты, потому что у них нет Y-аллосом.
Y-сцепленные расстройства чрезвычайно редки, но наиболее известные примеры обычно вызывают бесплодие. Воспроизведение в таких условиях возможно только путем предотвращения бесплодия с помощью медицинского вмешательства.
Митохондриальный
Этот тип наследования, также известный как материнское наследование, является самым редким и применим к 13 генам, кодируемым митохондриальная ДНК. Поскольку только яйцеклетки вносят вклад в митохондрии развивающегося эмбриона, только матери (пораженные) могут передавать состояние митохондриальной ДНК своим детям. Примером этого типа расстройства является Наследственная оптическая нейропатия Лебера.
Важно подчеркнуть, что подавляющее большинство митохондриальные заболевания (особенно когда симптомы развиваются в раннем возрасте) на самом деле вызваны ядерный ген дефект, так как митохондрии в основном развиваются немитохондриальной ДНК. Эти заболевания чаще всего возникают по аутосомно-рецессивному наследованию.[27]
Многофакторное расстройство
Генетические расстройства также могут быть сложными, многофакторными или полигенными, что означает, что они, вероятно, связаны с воздействием нескольких генов в сочетании с образом жизни и факторами окружающей среды. Многофакторные расстройства включают: сердечное заболевание и сахарный диабет. Хотя сложные расстройства часто группируются в семьях, они не имеют четкой наследственности. Это затрудняет определение риска для человека унаследовать или передать эти расстройства. Сложные расстройства также трудно изучать и лечить, поскольку конкретные факторы, вызывающие большинство этих расстройств, еще не определены. Исследования, направленные на определение причины сложных расстройств, могут использовать несколько методологических подходов для определения генотип –фенотип ассоциации. Один метод, генотип-первый подход, начинается с выявления генетических вариантов у пациентов и затем определения связанных клинических проявлений. Это противоположно более традиционному подходу, основанному на фенотипе, и может выявить причинные факторы, которые ранее были скрыты клиническими исследованиями. неоднородность, пенетрантность, и выразительность.
В родословной полигенные болезни имеют тенденцию «передаваться по наследству», но наследование не соответствует простым образцам, как в случае с Менделевский болезни. Но это не означает, что гены невозможно найти и изучить. Во многих из них также присутствует сильная экологическая составляющая (например, артериальное давление ).
- астма
- аутоиммунные заболевания Такие как рассеянный склероз
- раки
- цилиопатии
- волчья пасть
- сахарный диабет
- сердечное заболевание
- гипертония
- воспалительное заболевание кишечника
- Интеллектуальная недееспособность
- перепады настроения
- ожирение
- рефракционная ошибка
- бесплодие
Хромосомное расстройство
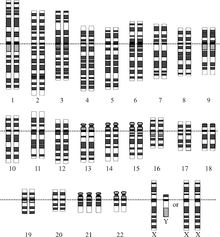
Хромосомное заболевание - это отсутствующая, лишняя или нерегулярная часть хромосомной ДНК. Это может быть из-за атипичного количества хромосом или структурной аномалии в одной или нескольких хромосомах. Примером этих нарушений является трисомия 21 (Синдром Дауна ), в котором есть лишняя копия хромосомы 21.
Диагностика
Из-за широкого спектра известных генетических нарушений диагноз широко варьируется и зависит от заболевания. Большинство генетических нарушений диагностируется при рождении или в раннем детстве, однако некоторые, например, болезнь Хантингтона, может ускользнуть от обнаружения, пока пациент не достигнет совершеннолетия.
Основные аспекты генетического расстройства основаны на наследовании генетического материала. С глубоким история семьи, можно предвидеть возможные расстройства у детей, которые направляют медицинских работников на специальные тесты в зависимости от расстройства и дают родителям возможность подготовиться к потенциальным изменениям образа жизни, предвидеть возможность мертворождение, или созерцать прекращение.[28] Пренатальная диагностика может обнаружить наличие характерных отклонений в развитии плода с помощью УЗИ, или обнаружить присутствие характерных веществ с помощью инвазивные процедуры которые включают введение зондов или игл в матку, например, амниоцентез.[29]
Прогноз
Не все генетические нарушения напрямую приводят к смерти; однако известных лекарств от генетических заболеваний нет. Многие генетические нарушения влияют на стадии развития, например: Синдром Дауна, в то время как другие приводят к чисто физическим симптомам, таким как мышечная дистрофия. Другие расстройства, такие как болезнь Хантингтона, не показывать никаких признаков до совершеннолетия. В активный период генетического нарушения пациенты в основном полагаются на поддержание или замедление деградации качество жизни и поддерживать пациента автономия. Это включает в себя физиотерапия, контроль над болью, и может включать в себя выбор Альтернативная медицина программы.
Уход

Лечение генетических заболеваний - это постоянная битва, в которой более 1800 генная терапия клинические испытания завершены, продолжаются или были одобрены во всем мире.[30] Несмотря на это, большинство вариантов лечения сосредоточены на лечении симптомов расстройств в попытке улучшить состояние пациента. качество жизни.
Генная терапия относится к форме лечения, при которой пациенту вводят здоровый ген. Это должно устранить дефект, вызванный дефектным геном, или замедлить прогрессирование заболевания. Основным препятствием была доставка генов в соответствующие клетки, ткани и органы, пораженные заболеванием. Как ввести ген в потенциально триллионы клеток, несущих дефектную копию? Этот вопрос был преградой между пониманием генетического нарушения и исправлением генетического нарушения.[31]
Эпидемиология
Примерно 1 из 50 человек страдает известным моногенным заболеванием, в то время как примерно 1 из 263 человек страдает от хромосомное заболевание.[6] Около 65% людей имеют проблемы со здоровьем в результате врожденных генетических мутаций.[6] Из-за значительного количества генетических нарушений примерно каждый 21 человек страдает генетическим заболеванием, классифицируемым как "редкий "(обычно определяется как поражение менее 1 из 2 000 человек). Большинство генетических нарушений редки сами по себе.[5][7] Известно более 6000 генетических заболеваний,[4] и новые генетические нарушения постоянно описываются в медицинской литературе.[5]
История
Самое раннее известное генетическое заболевание гоминид был в ископаемом виде Paranthropus robustus, более трети людей демонстрируют несовершенный амелогенез.[32]
Смотрите также
- FINDbase (база данных Частота наследственных заболеваний)
- Генетическая эпидемиология
- Список генетических нарушений
- Группы населения в биомедицине
- Менделирующая ошибка
Рекомендации
- ^ «Генетические заболевания». learn.genetics.utah.edu. Получено 2019-07-01.
- ^ Львов, Д .; Фаворова, О.О .; Фаворов, А. (2012). «Полигенный подход к изучению полигенных заболеваний». Acta Naturae. 4 (3): 59–71. Дои:10.32607/20758251-2012-4-3-59-71. ISSN 2075-8251. ЧВК 3491892. PMID 23150804.
- ^ Справка, Дом генетики. «Каковы различные способы наследования генетического заболевания?». Домашний справочник по генетике. Получено 2020-01-14.
- ^ а б "Статистика карты генов OMIM". www.omim.org. Получено 2020-01-14.
- ^ а б c d «Сирота: О редких болезнях». www.orpha.net. Получено 2020-01-14.
- ^ а б c d Кумар, Панкадж; Радхакришнан, Веселый; Chowdhary, Maksud A .; Джампьетро, Филипп Ф. (2001-08-01). «Распространенность и закономерности проявления генетических заболеваний в педиатрическом отделении неотложной помощи». Труды клиники Мэйо. 76 (8): 777–783. Дои:10.4065/76.8.777. ISSN 0025-6196.
- ^ а б Джексон, Мария; Маркс, Лия; May, Gerhard H.W .; Уилсон, Джоанна Б. (2018-12-03). «Генетическая основа болезни». Очерки биохимии. 62 (5): 643–723. Дои:10.1042 / EBC20170053. ISSN 0071-1365. ЧВК 6279436. PMID 30509934.
(рассчитано на основе «1 из 17» редких заболеваний и «80%» редких заболеваний являются генетическими)
- ^ «Генетика рака». www.medschool.lsuhsc.edu. Получено 2020-01-14.
- ^ "Запись OMIM № 144010 - ГИПЕРХОЛЕСТЕРОЛЕМИЯ, СЕМЕЙНАЯ, 2; FCHL2". www.omim.org. Получено 2019-07-01.
- ^ Simons, M .; Вальц, Г. (сентябрь 2006 г.). «Поликистоз почек: деление клеток без c (l) ue?». Kidney International. 70 (5): 854–864. Дои:10.1038 / sj.ki.5001534.
- ^ «Запись OMIM № 162200 - НЕЙРОФИБРОМАТОЗ, ТИП I; NF1». www.omim.org. Получено 2019-07-01.
- ^ Keane MG; Pyeritz RE (май 2008 г.). «Медицинское лечение синдрома Марфана». Тираж. 117 (21): 2802–13. Дои:10.1161 / CIRCULATIONAHA.107.693523. PMID 18506019.
- ^ Уокер Ф.О. (2007). "Болезнь Хантингтона". Ланцет. 369 (9557): 218–28 [221]. Дои:10.1016 / S0140-6736 (07) 60111-1. PMID 17240289.
- ^ «Запись OMIM № 603903 - СЕРПНО-КЛЕТОЧНАЯ АНЕМИЯ». www.omim.org. Получено 2019-07-01.
- ^ Уильямс Т. Н .; Обаро С. К. (2011). «Серповидно-клеточная анемия и заболеваемость малярией: история с двумя хвостами». Тенденции в паразитологии. 27 (7): 315–320. Дои:10.1016 / j.pt.2011.02.004. PMID 21429801.
- ^ Кулиев, Анвер; Верлинский, Юрий (2005). «Преимплантационная диагностика: реалистичный вариант для вспомогательной репродуктивной медицины и генетической практики». Curr. Мнение. Акушерство. Гинеколь. 17 (2): 179–83. Дои:10.1097 / 01.gco.0000162189.76349.c5. PMID 15758612.
- ^ Симчикова Д., Хенеберг П. (декабрь 2019 г.). «Уточнение прогнозов эволюционной медицины на основе клинических данных о проявлениях менделевских болезней». Научные отчеты. 9 (1): 18577. Дои:10.1038 / s41598-019-54976-4. ЧВК 6901466. PMID 31819097.CS1 maint: использует параметр авторов (связь)
- ^ а б Гриффитс, Энтони Дж. Ф .; Wessler, Susan R .; Кэрролл, Шон Б.; Добли, Джон (2012). «2: Наследование одного гена». Введение в генетический анализ (10-е изд.). Нью-Йорк: W.H. Фримен и компания. ISBN 978-1-4292-2943-2.
- ^ «Паттерны наследования для заболеваний одного гена». learn.genetics.utah.edu. Получено 2019-07-01.
- ^ Уэйд, Николас (29 января 2006 г.). «Японские ученые идентифицируют ген ушной серы». Нью-Йорк Таймс.
- ^ Yoshiura K; Киношита А; Исида Т; и другие. (Март 2006 г.). «SNP в гене ABCC11 является детерминантой типа ушной серы человека». Nat. Genet. 38 (3): 324–30. Дои:10,1038 / ng1733. PMID 16444273.
- ^ Миттон, Джеффри Б. (2002). «Гетерозиготное преимущество». eLS. Дои:10.1038 / npg.els.0001760. ISBN 0470016175.
- ^ Пулман Э.М., Гальвани А.П. (февраль 2007 г.). «Оценка кандидатов в агенты селективного давления при муковисцидозе». Журнал Королевского общества, Интерфейс. 4 (12): 91–8. Дои:10.1098 / rsif.2006.0154. ЧВК 2358959. PMID 17015291.
- ^ Allison AC (октябрь 2009 г.). «Генетический контроль устойчивости к малярии человека». Текущее мнение в иммунологии. 21 (5): 499–505. Дои:10.1016 / j.coi.2009.04.001. PMID 19442502.
- ^ Вульф, LI (1986). «Преимущество гетерозигот при фенилкетонурии». Американский журнал генетики человека. 38 (5): 773–5. ЧВК 1684820. PMID 3717163.
- ^ Уизералл, Д. Дж. (2015). «Талассемии: нарушения синтеза глобина». Гематология Вильямса (9e изд.). McGraw Hill Professional. п. 725. ISBN 9780071833011.
- ^ Нуссбаум, Роберт; Макиннес, Родерик; Уиллард, Хантингтон (2007). Томпсон и Томпсон Генетика в медицине. Филадельфия, Пенсильвания: Сондерс. С. 144, 145, 146. ISBN 9781416030805.
- ^ Милунский, Обри, изд. (2004). Генетические нарушения и плод: диагностика, профилактика и лечение (5-е изд.). Балтимор: Издательство Университета Джона Хопкинса. ISBN 978-0801879289.
- ^ «Диагностические исследования - Амниоцентез». Гарвардская медицинская школа. Архивировано из оригинал на 2008-05-16. Получено 2008-07-15.
- ^ Ginn, Samantha L .; Александр, Ян Э .; Эдельштейн, Майкл Л .; Abedi, Mohammad R .; Уиксон, Джо (февраль 2013 г.). «Клинические испытания генной терапии в мире до 2012 года - обновленная информация». Журнал генной медицины. 15 (2): 65–77. Дои:10.1002 / jgm.2698. PMID 23355455.
- ^ Верма, И. М. (22 августа 2013 г.). «Генная терапия, которая работает». Наука. 341 (6148): 853–855. Bibcode:2013Наука ... 341..853В. Дои:10.1126 / science.1242551. PMID 23970689.
- ^ «Вероятное генетическое происхождение точечной гипоплазии эмали на молярах Paranthropus robustus». ResearchGate. Получено 2019-03-09.
внешняя ссылка
| Классификация |
|---|
- Геномика общественного здравоохранения в CDC
- OMIM - онлайн-Менделирующее наследование в человеке, каталог человеческих генов и генетических нарушений
- Информационный центр по генетическим и редким заболеваниям (GARD) Управление редких заболеваний (ORD), Национальные институты здравоохранения (NIH)
- Национальный центр CDC по врожденным дефектам и порокам развития
- Информация о генетических заболеваниях из проекта "Геном человека"
- Глобальный проект генов, Организация по генетическим и редким заболеваниям
- Список генетических заболеваний - Genome.gov