Фенилкетонурия - Phenylketonuria
| Фенилкетонурия | |
|---|---|
| Другие имена | Дефицит фенилаланингидроксилазы, дефицит ПАУ, болезнь Фёллинга[1] |
 | |
| Фенилаланин | |
| Специальность | Медицинская генетика, педиатрия |
| Симптомы | Без лечения Интеллектуальная недееспособность, припадки, поведенческие проблемы, психические расстройства, затхлый запах[1] |
| Обычное начало | При рождении[2] |
| Типы | Классический, вариант[1] |
| Причины | Генетический (аутосомно-рецессивный )[1] |
| Диагностический метод | Программы скрининга новорожденных во многих странах[3] |
| Уход | Рацион питания мало продуктов, содержащих фенилаланин, специальные добавки[2] |
| Медикамент | Дигидрохлорид сапроптерина,[2] Pegvaliase[4] |
| Прогноз | Нормальное здоровье при лечении[5] |
| Частота | ~ 1 из 12 000 новорожденных[6] |
Фенилкетонурия (ФКУ) является врожденная ошибка обмена веществ что приводит к уменьшению метаболизм из аминокислота фенилаланин.[3] Без лечения ФКУ может привести к Интеллектуальная недееспособность, припадки, поведенческие проблемы и психические расстройства.[1] Это также может привести к появлению затхлого запаха и осветлению кожи.[1] Ребенок, рожденный от матери, которая плохо лечится от фенилкетонурии, может иметь проблемы с сердцем, маленькая голова, и низкий вес при рождении.[1]
Фенилкетонурия - генетическое заболевание. унаследованный от родителей человека.[1] Это связано с мутациями в ПАУ ген, что приводит к низкому уровню фермент фенилаланингидроксилаза.[1] Это приводит к накоплению диетического фенилаланина до потенциально токсичных уровней.[1] это аутосомно-рецессивный, что означает, что обе копии гена должны быть мутированы для развития состояния.[1] Существует два основных типа, классический PKU и вариант PKU, в зависимости от того, сохраняется ли какая-либо функция фермента.[1] Те, у кого есть одна копия мутировавшего гена, обычно не имеют симптомов.[1] Во многих странах программы скрининга новорожденных за болезнь.[3]
Лечение заключается в соблюдении диеты с низким содержанием фенилаланина и специальных добавок.[2] Младенцы должны использовать специальные формула с небольшим количеством грудное молоко.[2] Диету следует начинать как можно раньше после рождения и продолжать на всю жизнь.[2] Люди, которым поставлен ранний диагноз и соблюдают строгую диету, могут иметь нормальное здоровье и нормальную срок жизни.[5] Эффективность контролируют с помощью периодических анализов крови.[5] Лекарство дигидрохлорид сапроптерина может быть полезно в некоторых.[2]
Фенилкетонурия поражает примерно 1 из 12 000 детей.[6] Мужчины и женщины страдают одинаково.[7] Заболевание было обнаружено в 1934 г. Ивар Асбьёрн Фёллинг, когда важность диеты была определена в 1953 году.[8] Генная терапия, хотя и многообещающе, по состоянию на 2014 год требует гораздо большего изучения.[9]
Признаки и симптомы

Без лечения ФКУ может привести к Интеллектуальная недееспособность, припадки, поведенческие проблемы и психические расстройства.[1] Это также может привести к появлению затхлого запаха и осветлению кожи.[1] Ребенок, рожденный от матери, которая плохо лечится от фенилкетонурии, может иметь проблемы с сердцем, маленькая голова, и низкий вес при рождении.[1]
Поскольку организм матери способен расщеплять фенилаланин во время беременности, младенцы с фенилкетонурией при рождении нормальны. В то время болезнь не обнаруживается при физикальном обследовании, потому что повреждений еще не было. Скрининг новорожденных проводится для выявления заболевания и начала лечения до того, как будет нанесен какой-либо ущерб. Образец крови обычно берется пятка укол, как правило, выполняется через 2–7 дней после рождения. Этот тест может выявить повышенный уровень фенилаланина после одного или двух дней нормального кормления ребенка.[10][11]
Если ребенку не будет поставлен диагноз во время планового обследования новорожденных и не будет введена диета с ограничением фенилаланина, то со временем уровень фенилаланина в крови будет увеличиваться. Токсичные уровни фенилаланина (и недостаточные уровни тирозина) могут влиять на развитие ребенка и иметь необратимые последствия. Клинически заболевание может проявляться припадки, гипопигментация (чрезмерно светлые волосы и кожа) и "затхлый запах" пота и мочи ребенка (из-за фенилацетат, карбоновая кислота, получаемая при окислении фенилкетона). В большинстве случаев повторный тест следует сделать примерно в двухнедельном возрасте, чтобы проверить первоначальный тест и выявить любую фенилкетонурию, которая была изначально пропущена.[нужна цитата ]
Дети, не получающие лечения, часто не достигают вех в раннем развитии, у них развивается микроцефалия и наблюдается прогрессирующее нарушение церебральной функции. Гиперактивность, ЭЭГ аномалии, судороги и тяжелые неспособность к обучению являются серьезными клиническими проблемами в более позднем возрасте. Характерный «затхлый или мышечный» запах на коже, а также предрасположенность к экзема, сохраняются на протяжении всей жизни при отсутствии лечения.[нужна цитата ]
Повреждение головного мозга при отсутствии лечения фенилкетонурии в течение первых месяцев жизни необратимо. Очень важно очень тщательно контролировать диету младенцев с фенилкетонурией, чтобы мозг имел возможность нормально развиваться. У больных детей, которые выявляются при рождении и проходят лечение, гораздо реже развиваются неврологические проблемы, судороги и умственная отсталость (хотя такие клинические расстройства все еще возможны, включая астму, экзему, анемию, увеличение веса, почечную недостаточность, остеопороз, гастрит, пищевод и др.) почечная недостаточность, камни в почках и гипертония). Кроме того, серьезные депрессивные расстройства встречаются на 230% чаще, чем в контрольной группе; головокружение и головокружение возникают на 180% чаще; хроническая ишемическая болезнь сердца, астма, диабет и гастроэнтерит встречаются на 170% чаще; стрессовые и адаптационные расстройства встречаются на 160% чаще.[12][13] В целом, однако, результаты лечения людей от фенилкетонурии хорошие. У пролеченных людей могут вообще не быть обнаруживаемых физических, неврологических проблем или проблем развития.
Генетика

ФКУ - это аутосомно-рецессивный метаболический генетическое расстройство. Как аутосомно-рецессивное заболевание, две ФКУ аллели необходимы для того, чтобы человек испытал симптомы болезни. Чтобы ребенок унаследовал фенилкетонурию, и мать, и отец должны иметь и передавать дефектный ген.[14] Если оба родителя являются носителями фенилкетонурии, существует 25% вероятность того, что любой из их детей родится с этим заболеванием, вероятность 50%, что ребенок будет носителем, и вероятность 25%, что ребенок не разовьется и не будет носителем. за болезнь.[5]
ФКУ характеризуется гомозиготный или же составной гетерозиготный мутации в гене печеночного фермента фенилаланингидроксилаза (PAH), что делает его нефункциональным.[15]:541 Этот фермент необходим для метаболизма аминокислоты. фенилаланин (Phe) к аминокислоте тирозин (Тюр). Когда активность ПАУ снижается, фенилаланин накапливается и превращается в фенилпируват (также известный как фенилкетон), который можно обнаружить в моча.[16]
Носители одного аллеля PKU не проявляют симптомов заболевания, но, по-видимому, в некоторой степени защищены от грибкового токсина. охратоксин А.[17] Этим объясняется устойчивость аллеля в определенных популяциях, поскольку он дает селективное преимущество - другими словами, быть гетерозигота выгодна.[18]
Ген PAH расположен на хромосома 12 в полосах 12q22-q24.2.[19] По состоянию на 2000 год в гене PAH было обнаружено около 400 болезнетворных мутаций. Это пример аллельного генетическая гетерогенность.[5]
Патофизиология
Когда фенилаланин (Phe) не может быть метаболизирован организмом, типичная диета, которая была бы здоровой для людей без фенилкетонурии, вызывает накопление в крови аномально высоких уровней Phe, что токсично для мозга. Если не лечить, к осложнениям ФКУ относятся тяжелая умственная отсталость, нарушения функции головного мозга, микроцефалия, расстройства настроения, нерегулярное двигательное функционирование и поведенческие проблемы, такие как Синдром дефицита внимания и гиперактивности, а также физические симптомы, такие как «затхлый» запах, экзема и необычно светлый цвет кожи и волос.[нужна цитата ]
Классическая ФКУ
Классическая ФКУ и ее менее тяжелые формы «легкая ФКУ» и «легкая гиперфенилаланинемия» вызываются мутировавшим геном фермент фенилаланингидроксилаза (ПАУ), который превращает аминокислоту фенилаланин («Phe») в другие важные соединения в организме, в частности тирозин. Тирозин является условно незаменимым аминокислота для пациентов с фенилкетонурией, потому что без ПАУ она не может вырабатываться в организме за счет распада фенилаланина. Тирозин необходим для выработки нейротрансмиттеров, таких как адреналин, норадреналин и дофамин.[20]
Дефицит ПАУ вызывает целый спектр заболеваний, включая классическую фенилкетонурию (ФКУ) и легкую гиперфенилаланинемию (также известную как «гиперфекция»).[21] или «мягкий HPA»), менее серьезное накопление фенилаланина. По сравнению с пациентами с классической фенилкетонурией, пациенты с «гиперфеем» обладают большей активностью фермента ПАУ и способны переносить большее количество фенилаланина в своем рационе. Без диетического вмешательства у пациентов с легкой формой HPA уровень Phe в крови выше, чем у пациентов с нормальной активностью PAH. В настоящее время нет международного консенсуса в отношении определения легкого HPA, однако он чаще всего диагностируется при уровне Phe в крови от 2 до 6 мг / дл.[22]
Фенилаланин - большая нейтральная аминокислота (LNAA). LNAA конкурируют за транспортировку через гематоэнцефалический барьер (BBB) через большой переносчик нейтральных аминокислот (LNAAT). Если фенилаланин находится в избытке в крови, он насыщает транспортер. Избыточные уровни фенилаланина имеют тенденцию к снижению уровней других LNAA в головном мозге. Поскольку эти аминокислоты необходимы для синтеза белка и нейромедиаторов, накопление Phe препятствует развитию мозг, вызывая Интеллектуальная недееспособность.[23]
Недавние исследования показывают, что нейрокогнитивные, психосоциальные, качество жизни, рост, питание, патология костей несколько неоптимальны даже для пациентов, которые проходят лечение и поддерживают уровень Phe в целевом диапазоне, если их диета не дополняется другими аминокислотами.[24]
Классическая фенилкетонурия поражает миелинизацию и тракты белого вещества у нелеченных младенцев; это может быть одной из основных причин неврологических проблем, связанных с фенилкетонурией. Различия в развитии белого вещества наблюдаются при магнитно-резонансная томография. Также могут быть обнаружены аномалии серого вещества,[25] особенно в моторной и домоторной коре, таламусе и гиппокампе.[нужна цитата ]
Недавно было высказано предположение, что ФКУ может напоминать амилоид заболевания, такие как болезнь Альцгеймера и болезнь Паркинсона, из-за образования токсичных амилоидоподобных комплексов фенилаланина.[26]
Другие мутации, не связанные с ПАУ, также могут вызывать ФКУ.[нужна цитата ]
Гиперфенилаланинемия с дефицитом тетрагидробиоптерина
Более редкой формой гиперфенилаланинемии является дефицит тетрагидробиоптерина, который возникает, когда фермент ПАУ в норме, и обнаружен дефект в биосинтезе или рециркуляции кофактор тетрагидробиоптерин (BH4).[27] BH4 необходим для правильной активности фермента ПАУ, и это кофермент можно дополнить как лечение. Те, кто страдает этой формой гиперфенилаланинемии, могут иметь дефицит тирозина (который создается из фенилаланина с помощью ПАУ), и в этом случае лечение заключается в добавлении тирозина для компенсации этого дефицита.[нужна цитата ]
Уровни дофамин можно использовать для различения этих двух типов. Тетрагидробиоптерин требуется для преобразования Phe в Tyr и требуется для преобразования Tyr в L-ДОПА через фермент тирозингидроксилаза. L-ДОПА, в свою очередь, преобразуется в дофамин. Низкий уровень дофамина приводит к высокому уровню пролактин. Напротив, при классической ФКУ (без участия дигидробиоптерина) уровни пролактина будут относительно нормальными.[нужна цитата ]
Дефицит тетрагидробиоптерина может быть вызван дефектами четырех генов. Они известны как HPABH4A, HPABH4B, HPABH4C и HPABH4D.[28]
Метаболические пути

Фермент фенилаланингидроксилаза обычно преобразует аминокислота фенилаланин в аминокислоту тирозин. Если эта реакция не происходит, фенилаланин накапливается и тирозин недостаточен. Избыточный фенилаланин может метаболизироваться в фенилкетоны второстепенным путем: трансаминаза путь с глутамат. Метаболиты включают фенилацетат, фенилпируват и фенэтиламин.[29] Повышенные уровни фенилаланина в крови и обнаружение фенилкетонов в моче являются диагностическими, однако у большинства пациентов диагноз ставится при скрининге новорожденных.
Скрининг
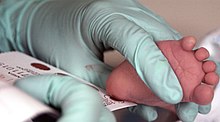
ФКУ обычно включается в обследование новорожденных панель многих стран с различными методами обнаружения. Большинство детей в развитых странах проходят скрининг на ФКУ вскоре после рождения.[30] Скрининг на ФКУ проводится с помощью анализа ингибирования бактерий (Тест Гатри ), иммуноанализы с использованием флуорометрического или фотометрического детектирования или измерения аминокислот с использованием тандемная масс-спектрометрия (МС / МС). Измерения, проведенные с помощью МС / МС, определяют концентрацию Phe и отношение Phe к тирозин, соотношение будет повышено в ФКУ.[31]
Уход
ФКУ не излечима. Однако, если ФКУ диагностируется достаточно рано, пораженный новорожденный может расти с нормальным развитием мозга, управляя и контролируя уровни фенилаланина («Phe») с помощью диеты или комбинации диеты и лекарств.[нужна цитата ]
Рацион питания
У людей, которые с рождения соблюдают предписанное диетическое лечение, симптомы могут отсутствовать. Их ФКУ можно будет обнаружить только с помощью анализа крови. Люди должны придерживаться специальной диеты с низким содержанием Phe для оптимального развития мозга. Поскольку Phe необходим для синтеза многих белков, он необходим для соответствующего роста, но его уровни необходимо строго контролировать.[нужна цитата ]
Для людей, не страдающих фенилкетонурией, Институт медицины США рекомендовал по крайней мере 33 мг / кг массы тела / день фенилаланина плюс тирозин для взрослых от 19 лет и старше.[32] Для людей с фенилкетонурией рекомендуется для детей в возрасте до 10 лет от 200 до 500 мг / сут; для детей старшего возраста и взрослых от 220 до 1200 мг / сут. Где находится диапазон, зависит от массы тела и возраста, а также от мониторинга концентрации в крови.[33]
Оптимальные диапазоны здоровья (или "целевые диапазоны") от 120 до 360 мкмоль / л или, что эквивалентно от 2 до 6 мг / дл, и нацелено на достижение по крайней мере в течение первых 10 лет,[34] чтобы мозг мог нормально развиваться.
Возраст, в котором люди с фенилкетонурией могут безопасно отказаться от диеты, является предметом некоторых дискуссий. Диету нужно соблюдать как минимум до восьми-десяти лет. Некоторые данные подтверждают, что прекращение приема через 10 лет, поскольку обычная диета после этого не оказывает отрицательного воздействия.[35] Однако одно исследование показало временные пагубные эффекты при отказе от диеты.[36] Нет доказательств необратимого повреждения мозга у людей, которые отказались от диеты в зрелом возрасте.[37][35] В случае легких нейрокогнитивных нарушений показано возобновление диеты.
Диета требует ограничения или исключения продуктов с высоким содержанием Phe, таких как соевые бобы, яичные белки, креветка, куриная грудка, спирулина, кресс-салат, рыбы, орехи, рак, Омар, тунец, индюк, бобовые и с низким содержанием жира творог.[38] Крахмалистые продукты, такие как картофель и кукуруза обычно приемлемы в контролируемых количествах, но необходимо контролировать количество Phe, потребляемого из этих продуктов. Дневник питания обычно ведется для записи количества Phe, потребляемого с каждым приемом пищи, перекусом или напитком. Для расчета количества Phe в пище по содержанию белка, указанному на этикетке с информацией о пищевой ценности, можно использовать систему «обмена». Заменители «лечебной пищи» с низким содержанием белка часто используются вместо обычных хлеб, макароны и другие продукты на основе зерна, которые содержат значительное количество Phe. Многие фрукты и овощи содержат меньше Phe, и их можно есть в больших количествах. Младенцы по-прежнему могут находиться на грудном вскармливании, чтобы обеспечить все преимущества грудного молока, но количество также необходимо контролировать, и потребуются добавки для недостающих питательных веществ. Подсластитель аспартам, присутствующий во многих диетических продуктах и безалкогольных напитках, также следует избегать, поскольку аспартам метаболизируется в фенилаланин.[нужна цитата ]
Разные люди могут переносить разное количество Phe в своем рационе. Регулярные анализы крови используются для определения влияния потребления Phe с пищей на уровень Phe в крови.
Пищевые добавки
Людям с фенилкетонурией (начиная с младенчества) обычно назначают дополнительные формулы «заменителя белка», чтобы обеспечить аминокислоты и другие необходимые питательные вещества, которых в противном случае не хватало бы в диете с низким содержанием фенилаланина. Тирозин, который обычно получают из фенилаланина и который необходим для нормальной работы мозга, обычно дополняется. Употребление формул, заменяющих протеин, может снизить уровень фенилаланина, вероятно, потому, что он останавливает процесс протеина. катаболизм от высвобождения Phe, хранящегося в мышцах и других тканях, в кровь. Многие пациенты с фенилкетонурией имеют самый высокий уровень Phe после периода голодания (например, в течение ночи), потому что голодание вызывает катаболизм.[39] Диета с низким содержанием фенилаланина, но не содержащая заменителей белка, также может не снизить уровень Phe в крови, поскольку диета с недостаточным питанием также может вызвать катаболизм. По всем этим причинам рецептурная формула является важной частью лечения пациентов с классической фенилкетонурией.[нужна цитата ]
Предварительные данные подтверждают диетические добавки с большими нейтральными аминокислотами (LNAA).[40] LNAA (например, лея, тыр, trp, встретились, его, ile, вал, чт ) может конкурировать с phe за специфические белки-носители, которые транспортируют LNAA через слизистую оболочку кишечника в кровь и через гематоэнцефалический барьер в мозг. Его использование действительно показано только взрослым, которые не соблюдают соответствующую диету.[3]
Другой интересной стратегией лечения является гликомакропептид казеина (CGMP), который представляет собой молочный пептид, естественно не содержащий Phe в чистом виде.[41] CGMP может заменять основную часть свободных аминокислот в диете PKU и обеспечивает несколько полезных питательных эффектов по сравнению со свободными аминокислотами. Тот факт, что CGMP является пептидом, гарантирует, что скорость абсорбции его аминокислот увеличивается по сравнению со свободными аминокислотами и, таким образом, приводит к улучшенному удержанию белка.[42] и повышенное сытость[43] по сравнению со свободными аминокислотами. Еще одно важное преимущество CGMP - значительно улучшенный вкус.[42] когда CGMP заменяет часть свободных аминокислот, и это может помочь улучшить соблюдение диеты для лечения фенилкетонурии.
Кроме того, CGMP содержит большое количество Phe-снижающих LNAA, которое составляет около 41 г на 100 г белка.[41] и, следовательно, поможет поддерживать уровни phe в плазме в целевом диапазоне.
Заменители ферментов
В 2018 году FDA одобрило заменитель фермента под названием Pegvaliase который метаболизирует фенилаланин.[4] Он предназначен для взрослых, которым другие методы лечения неэффективны.[4]
Тетрагидробиоптерин (BH4) (кофактор для окисление фенилаланина) при приеме внутрь может уменьшить кровь уровни этой аминокислоты у некоторых людей.[44][45] Однако у большинства людей с «классической» последовательностью мутаций пользы будет мало или никакой.[9]
Матери
Для женщин, больных фенилкетонурией, для здоровья детей важно поддерживать низкий уровень Phe до и во время беременности.[46] Хотя развивающийся плод может быть носителем только гена PKU, внутриутробная среда может иметь очень высокие уровни фенилаланина, который может проникать через плаценту. В результате у ребенка могут развиться врожденные пороки сердца, задержка роста, микроцефалия и умственная отсталость.[47] Сами женщины, страдающие фенилкетонурией, не подвергаются риску дополнительных осложнений во время беременности.[нужна цитата ]
В большинстве стран женщинам с фенилкетонурией, желающим иметь детей, рекомендуется снизить уровень Phe в крови (обычно до 2–6 мг / дл) до того, как они забеременеют, и тщательно контролировать их уровни на протяжении всей беременности. Это достигается за счет проведения регулярных анализов крови и очень строгого соблюдения диеты, как правило, под ежедневным наблюдением специалиста-диетолога-метаболиста. Во многих случаях, когда печень плода начинает нормально развиваться и вырабатывать ПАУ, уровень Phe в крови матери будет падать, что требует увеличения потребления, чтобы оставаться в безопасном диапазоне 2–6 мг / дл. В результате суточное потребление Phe матерью может удвоиться или даже утроиться к концу беременности. Когда уровень Phe в крови матери падает ниже 2 мг / дл, отдельные сообщения указывают на то, что матери могут страдать от побочных эффектов, включая головные боли, тошноту, выпадение волос и общее недомогание. Когда низкие уровни фенилаланина поддерживаются на протяжении всей беременности, нет повышенного риска врожденных дефектов по сравнению с ребенком, рожденным от матери, не болеющей фенилкетонурией.[48]
Эпидемиология
| Страна | Заболеваемость |
|---|---|
| Австралия | 1 из 10 000[49] |
| Бразилия | 1 из 8690 |
| Канада | 1 из 22 000[49] |
| Китай | 1 из 17 000[49] |
| Чехословакия | 1 из 7000[49] |
| Дания | 1 из 12 000[49] |
| Финляндия | 1 из 200 000[49] |
| Франция | 1 из 13 500[49] |
| Индия | 1 из 18 300 |
| Ирландия | 1 из 4500[50] |
| Италия | 1 из 17 000[49] |
| Япония | 1 из 125 000[49] |
| Корея | 1 из 41 000[51] |
| Нидерланды | 1 из 18 000[52] |
| Норвегия | 1 из 14 500[49] |
| Филиппины | 1 из 102 000[53] |
| Польша | 1 из 8000[52] |
| Шотландия | 1 из 5300[49] |
| Испания | 1 из 20 000[52] |
| Швеция | 1 из 20 000[52] |
| индюк | 1 из 2600[49] |
| объединенное Королевство | 1 из 10 000[52] |
| Соединенные Штаты | 1 из 25 000[54] |
Среднее количество новых случаев фенилкетонурии варьируется в разных популяциях человека. Кавказцы в Соединенных Штатах страдают от этого заболевания в размере 1 из 10 000.[55] В Турции самый высокий зарегистрированный уровень в мире - 1 случай на 2600 рождений, в то время как в таких странах, как Финляндия и Япония, показатели чрезвычайно низкие - менее одного случая ФКУ на 100 000 рождений. В исследовании 1987 г. из Словакии сообщается о Рома популяция с чрезвычайно высокой заболеваемостью фенилкетонурией (один случай из 40 рождений) из-за обширного инбридинга.[56] Это самая распространенная проблема метаболизма аминокислот в Соединенном Королевстве.[нужна цитата ]
История
До того, как были выяснены причины ФКУ, ФКУ вызывала тяжелую инвалидность у большинства людей, унаследовавших соответствующие мутации. Автор, лауреат Нобелевской и Пулитцеровской премий Перл С. Бак у нее была дочь по имени Кэрол, которая жила с фенилкетонурией до того, как стало доступно лечение, и написала отчет о его эффектах в книге под названием Ребенок, который никогда не вырос.[57] Многие нелеченные пациенты с фенилкетонурией, родившиеся до широкомасштабного скрининга новорожденных, все еще живы, в основном в домах / учреждениях с зависимым проживанием.[58]
Фенилкетонурия была обнаружена норвежский язык врач Ивар Асбьёрн Фёллинг в 1934 г.[59] когда он заметил, что гиперфенилаланинемия (HPA) была связана с умственной отсталостью. В Норвегии это заболевание известно как болезнь Феллинга, названная в честь первооткрывателя.[60] Фёллинг был одним из первых врачей, применивших подробный химический анализ к изучению болезней.
В 1934 г. Рикшоспиталет Фёллинг увидел молодую женщину по имени Борни Эгеланн. У нее было двое детей, Лив и Даг, которые были нормальными при рождении, но впоследствии у них развились умственные нарушения. Когда Дагу было около года, мать заметила сильный запах его мочи. Фёллинг взял у детей образцы мочи и после многих тестов обнаружил, что вещество, вызывающее запах в моче, - фенилпировиноградная кислота. Он пришел к выводу, что у детей был избыток фенилпировиноградной кислоты в моче, и это состояние было названо фенилкетонурией (ФКУ).[16]
Его тщательный анализ мочи двух пострадавших братьев и сестер привел к тому, что он попросил многих врачей недалеко от Осло проверить мочу других пострадавших пациентов. Это привело к открытию того же вещества, которое он обнаружил у восьми других пациентов. Он провел тесты и обнаружил реакции, которые привели к бензальдегид и бензойная кислота, что привело его к выводу, что соединение содержит бензол звенеть. Дальнейшее тестирование показало температура плавления быть таким же, как фенилпировиноградная кислота, что указывало на то, что вещество было в моче.[нужна цитата ]
В 1954 г. Хорст Бикель, Эвелин Хикманс и Джон Джеррард опубликовали статью, в которой описали, как они создали диету с низким содержанием фенилаланин и пациент поправился.[61] Бикель, Джеррард и Хикманс были награждены Медаль Джона Скотта в 1962 г. за их открытие.[61]
Фенилкетонурия была первым заболеванием, которое обычно диагностируется через широко распространенные обследование новорожденных. Роберт Гатри представила скрининговый тест новорожденных на ФКУ в начале 1960-х годов.[62] Зная, что ФКУ можно обнаружить до того, как проявятся симптомы и начнется лечение, скрининг был быстро принят во всем мире. Ирландия была первой страной, которая ввела национальную программу скрининга в феврале 1966 года.[63], Австрия также начала показ в 1966 году.[64] и Англия в 1968 году.[65]
В 2017 году были опубликованы Европейские рекомендации.[66] Их призывали такие организации пациентов, как Европейское общество фенилкетонурии и родственных заболеваний, рассматриваемых как фенилкетонурия.[67][68] Они были встречены критиками.[69]
Этимология и произношение
Слово фенилкетонурия использует комбинирование форм из фенил + кетон + -урия; это произносится /ˌжяпаɪлˌkятəˈНью-Джерсиʊərяə,ˌжɛп-,-пɪл-,-пəl-,-тoʊ-/[70][71].
Исследование
Другие методы лечения в настоящее время исследуются, в том числе генная терапия.
Биомарин в настоящее время проводит клинические испытания для изучения PEG-PAL (пегилированный рекомбинантный фенилаланин аммиаклиаза или «PAL») представляет собой заместительную ферментативную терапию, при которой отсутствующий фермент PAH заменяется аналогичным ферментом, который также расщепляет Phe. PEG-PAL сейчас находится в фазе 2 клинической разработки.[72]
Смотрите также
- Гиперфенилаланемия
- Лофеналак
- Дефицит тетрагидробиоптерина
- Цветы для Алджернона, который показывает персонажа с фенилкетонурией
Рекомендации
- ^ а б c d е ж грамм час я j k л м п о п «фенилкетонурия». Домашний справочник по генетике. 8 сентября 2016 г. В архиве из оригинала 27 июля 2016 г.. Получено 12 сентября 2016.
- ^ а б c d е ж грамм «Каковы общие методы лечения фенилкетонурии (ФКУ)?». NICHD. 2013-08-23. В архиве из оригинала 5 октября 2016 г.. Получено 12 сентября 2016.
- ^ а б c d Аль Хафид Н., Христодулу Дж. (Октябрь 2015 г.). «Фенилкетонурия: обзор текущих и будущих методов лечения». Переводческая педиатрия. 4 (4): 304–17. Дои:10.3978 / j.issn.2224-4336.2015.10.07. ЧВК 4728993. PMID 26835392.
- ^ а б c «Сообщения для прессы - FDA одобряет новый метод лечения фенилкетонурии, редкого и серьезного генетического заболевания». www.fda.gov. Получено 9 декабря 2018.
- ^ а б c d е "Заявление конференции по развитию консенсуса национальных институтов здравоохранения Фенилкетонурия: скрининг и лечение". NICHD. 16–18 октября 2000 г. В архиве из оригинала 5 октября 2016 г.. Получено 12 сентября 2016.
- ^ а б Бернштейн Л. Е., Рор Ф, Хельм Дж. Р. (2015). Управление питанием при наследственных метаболических заболеваниях: уроки метаболического университета. Springer. п. 91. ISBN 9783319146218. В архиве из оригинала от 11.09.2017.
- ^ Маркданте К., Клигман Р.М. (2014). Нельсон: основы педиатрии (7-е изд.). Elsevier Health Sciences. п. 150. ISBN 9780323226981. В архиве из оригинала от 11.09.2017.
- ^ Кальтер Х (2010). Тератология в двадцатом веке плюс десять. Springer Science & Business Media. С. 89–92. ISBN 9789048188208. В архиве из оригинала от 11.09.2017.
- ^ а б Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N и др. (Июнь 2014 г.). «Научная обзорная конференция по фенилкетонурии: состояние науки и будущие потребности в исследованиях». Молекулярная генетика и метаболизм. 112 (2): 87–122. Дои:10.1016 / j.ymgme.2014.02.013. PMID 24667081.
- ^ «Тест на фенилкетонурию (ФКУ)». HealthLink BC. Получено 28 августа, 2020.
- ^ Берри С.А., Браун С., Грант М., Грин С.Л., Юреки Э., Кох Дж. И др. (Август 2013). «Скрининг новорожденных 50 лет спустя: проблемы доступа, с которыми сталкиваются взрослые с ФКУ». Генетика в медицине. 15 (8): 591–9. Дои:10.1038 / gim.2013.10. ЧВК 3938172. PMID 23470838.
- ^ Бертон Б.К., Джонс К.Б., Седербаум С., Рор Ф., Вайсбрен С., Ирвин Д.Е.; и другие. (2018). «Распространенность коморбидных состояний среди взрослых пациентов с фенилкетонурией». Мол Генет Метаб. 125 (3): 228–234. Дои:10.1016 / j.ymgme.2018.09.006. PMID 30266197.CS1 maint: несколько имен: список авторов (связь)
- ^ Trefz KF, Muntau AC, Kohlscheen KM, Altevers J, Jacob C, Braun S; и другие. (2019). «Клиническое бремя болезни у пациентов с фенилкетонурией (ФКУ) и сопутствующими заболеваниями - ретроспективное исследование данных о страховых выплатах в Германии». Орфанет Дж. Редкие Диск.. 14 (1): 181. Дои:10.1186 / s13023-019-1153-у. ЧВК 6647060. PMID 31331350.CS1 maint: несколько имен: список авторов (связь)
- ^ «Фенилкетонурия (ФКУ) - симптомы и причины». Клиника Майо. Получено 2020-03-10.
- ^ Джеймс У.Д., Бергер Т.Г. (2006). Кожные болезни Эндрюса: клиническая дерматология. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ^ а б Гонсалес Дж., Уиллис М.С. (февраль 2010 г.). "Ивар Асбьорн Фёллинг". Лабораторная медицина. 41 (2): 118–119. Дои:10.1309 / LM62LVV5OSLUJOQF.
- ^ Вульф Л.И. (май 1986 г.). «Преимущество гетерозигот при фенилкетонурии». Американский журнал генетики человека. 38 (5): 773–5. ЧВК 1684820. PMID 3717163.
- ^ Льюис Р. (1997). Генетика человека. Чикаго, Иллинойс: Wm. К. Браун. С. 247–248. ISBN 978-0-697-24030-9.
- ^ Розенберг Р.Н., Барчи Р.Л., DiMauro S, Прусинер СБ, Nestler EJ (2003). Молекулярно-генетические основы неврологических и психиатрических заболеваний. Баттерворт-Хайнеманн. п. 820. ISBN 9780750673600.
- ^ «Фенилкетонурия». Линия здоровья. 20 августа 2012 г. В архиве с оригинала от 30 декабря 2014 г.
- ^ Regier DS, Грин CL. Дефицит фенилаланингидроксилазы. 10 января 2000 г. [Обновлено 5 января 2017 г.. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2020 гг.]
- ^ де ла Парра А., Гарсия М.И., Вайсбрен С.Е., Корнехо В., Райманн Э. (декабрь 2015 г.). «Когнитивные функции при легкой гиперфенилаланинемии». Отчеты по молекулярной генетике и метаболизму. 5: 72–75. Дои:10.1016 / j.ymgmr.2015.10.009. ЧВК 5471391. PMID 28649547.
- ^ Пиц Дж., Крайс Р., Рупп А., Маятепек Е., Рейтинг D, Бош С., Бремер Х. Дж. (1999). «Крупные нейтральные аминокислоты блокируют транспорт фенилаланина в ткани мозга у пациентов с фенилкетонурией». Журнал клинических исследований. 103 (8): 1169–1178. Дои:10.1172 / JCI5017. ЧВК 408272. PMID 10207169.
- ^ Эннс Г.М., Кох Р., Брамм В., Блейкли Е., Сутер Р., Юреки Е. (1 октября 2010 г.). «Неоптимальные исходы у пациентов с ФКУ, получавших лечение только с помощью диеты на ранней стадии: пересмотр доказательств» Молекулярная генетика и метаболизм. 101 (2–3): 99–109. Дои:10.1016 / j.ymgme.2010.05.017. PMID 20678948.
- ^ Террибилли Д., Шауфельбергер М.С. (10 мая 2020 г.). «Возрастные изменения объема серого вещества в головном мозге в зрелом возрасте». Нейробиология старения. 32 (2–6): 354–368. Дои:10.1016 / j.neurobiolaging.2009.02.008. ЧВК 3004040. PMID 19282066.
- ^ Адлер-Абрамович Л., Вакс Л., Карни О., Трудлер Д., Магно А., Кафлиш А., Френкель Д., Газит Е. (август 2012 г.). «Сборка фенилаланина в токсичные фибриллы предполагает амилоидную этиологию фенилкетонурии». Природа Химическая Биология. 8 (8): 701–6. Дои:10.1038 / nchembio.1002. PMID 22706200.
- ^ Сёртиз Р., Блау Н. (2000). «Нейрохимия фенилкетонурии». Европейский журнал педиатрии. 169: S109 – S113. Дои:10.1007 / PL00014370. PMID 11043156. S2CID 26196359.
- ^ Онлайн-менделевское наследование в человеке (OMIM): 261640
- ^ Михалс К., Маталон Р. (1985). «Метаболиты фенилаланина, концентрация внимания и гиперактивность». Американский журнал клинического питания. 42 (2): 361–5. Дои:10.1093 / ajcn / 42.2.361. PMID 4025205.
- ^ Персонал клиники Мэйо (2007-12-20). «Фенилкетонурия (ФКУ)». Клиника Майо. В архиве из оригинала 17.03.2008. Получено 2008-03-13.
- ^ Сарафоглу К., Хоффманн Г.Ф., Рот К.С. (ред.). Детская эндокринология и врожденные ошибки обмена веществ. Нью-Йорк: McGraw Hill Medical. п. 26.
- ^ Институт медицины (2002). «Белок и аминокислоты». Нормы потребления энергии, углеводов, клетчатки, жиров, жирных кислот, холестерина, белков и аминокислот с пищей. Вашингтон, округ Колумбия: The National Academies Press. С. 589–768.
- ^ Маклеод Э.Л., Ней Д.М. (2010). «Управление питанием при фенилкетонурии». Анналес Нестле (англ. Ред.). 68 (2): 58–69. Дои:10.1159/000312813. ЧВК 2901905. PMID 22475869.
- ^ Глава 55, страница 255 В архиве 2016-05-11 в Wayback Machine в:Берман, Ричард Э .; Клигман, Роберт; Нельсон, Уолдо Э .; Карен Маркданте; Дженсон, Хэл Б. (2006). Нельсон основы педиатрии. Elsevier / Saunders. ISBN 978-1-4160-0159-1.
- ^ а б «Политика ввода в клиническую эксплуатацию: сапроптерин (Куван®) для лечения фенилкетонурии: использование во время беременности» (PDF). Апрель 2013. с. 4. Получено 25 марта 2018.
- ^ "Национальные новости ФКУ: Взрослые". pkunews.org. В архиве из оригинала от 07.03.2015.
- ^ «Фенилкетонурия». nhs.uk. 19 октября 2017 г.. Получено 28 августа, 2020.
- ^ «Продукты с самым высоким содержанием фенилаланина». self.com. В архиве из оригинала от 05.05.2015.
- ^ Макдональд А., Райланс Г. В., Асплин Д., Холл СК, Бут И. В. (1998). «Предсказывает ли один фенилаланин в плазме качество контроля фенилкетонурии?». Архив детских болезней. 78 (2): 122–6. Дои:10.1136 / adc.78.2.122. ЧВК 1717471. PMID 9579152.
- ^ ван Спронсен Ф.Дж., де Гроот М.Дж., Хуксма М., Рейнгуд Д.Д., ван Рейн М. (декабрь 2010 г.). «Большие нейтральные аминокислоты в лечении ФКУ: от теории к практике». Журнал наследственных метаболических заболеваний. 33 (6): 671–6. Дои:10.1007 / s10545-010-9216-1. ЧВК 2992655. PMID 20976625.
- ^ а б Etzel MR (апрель 2004 г.). «Производство и использование фракций молочного белка». Журнал питания. 134 (4): 996S – 1002S. Дои:10.1093 / jn / 134.4.996S. PMID 15051860.
- ^ а б van Calcar SC, MacLeod EL, Gleason ST, Etzel MR, Clayton MK, Wolff JA, Ney DM (апрель 2009 г.). «Улучшенное питание при фенилкетонурии с помощью диеты, содержащей гликомакропептид, по сравнению с аминокислотами». Американский журнал клинического питания. 89 (4): 1068–77. Дои:10.3945 / ajcn.2008.27280. ЧВК 2667457. PMID 19244369.
- ^ МакЛауд Э.Л., Клейтон М.К., ван Калькар СК, Ней Д.М. (август 2010 г.). «Завтрак с гликомакропептидом по сравнению с аминокислотами подавляет уровень грелина в плазме у людей с фенилкетонурией». Молекулярная генетика и метаболизм. 100 (4): 303–8. Дои:10.1016 / j.ymgme.2010.04.003. ЧВК 2906609. PMID 20466571.
- ^ Бертон Б.К., Кар С., Киркпатрик П. (2008). «Сапроптерин». Обзоры природы Drug Discovery. 7 (3): 199–200. Дои:10.1038 / nrd2540.
- ^ Михалс-Маталон К. (февраль 2008 г.). «Дигидрохлорид сапроптерина, 6-R-L-эритро-5,6,7,8-тетрагидробиоптерин в лечении фенилкетонурии». Заключение эксперта по исследуемым препаратам. 17 (2): 245–51. Дои:10.1517/13543784.17.2.245. PMID 18230057. S2CID 207475494.
- ^ Ли PJ, Ridout D, Уолтер JH, Кокберн F (2005). «Материнская фенилкетонурия: отчет из реестра Соединенного Королевства 1978–97». Архив детских болезней. 90 (2): 143–146. Дои:10.1136 / adc.2003.037762. ЧВК 1720245. PMID 15665165.
- ^ Роуз Б., Азен С., Кох Р., Маталон Р., Хэнли В., де ла Круз Ф., Трефз Ф., Фридман Э., Шифрин Н. (1997). «Совместное исследование материнской фенилкетонурии (MPKUCS) у потомства: лицевые аномалии, пороки развития и ранние неврологические последствия». Американский журнал медицинской генетики. 69 (1): 89–95. Дои:10.1002 / (SICI) 1096-8628 (19970303) 69: 1 <89 :: AID-AJMG17> 3.0.CO; 2-K. PMID 9066890.
- ^ lsuhsc.edu В архиве 2008-04-08 на Wayback Machine Генетика и семьи Луизианы
- ^ а б c d е ж грамм час я j k л Уильямс Р.А., Mamotte CD, Бернетт-младший (февраль 2008 г.). «Фенилкетонурия: врожденная ошибка метаболизма фенилаланина». Клинический биохимик. Отзывы. 29 (1): 31–41. ЧВК 2423317. PMID 18566668.
- ^ DiLella AG, Kwok SC, Ledley FD, Marvit J, Woo SL (1986). «Молекулярная структура и полиморфная карта гена фенилаланингидроксилазы человека». Биохимия. 25 (4): 743–749. Дои:10.1021 / bi00352a001. PMID 3008810.
- ^ Ли Д.Х., Ку С.К., Ли К.С., Ён ЙДж, О ХД, Ким С.В., Ли С.Дж., Ким С.С., Ли Джи, Чо И, Юнг СК (2004). «Молекулярные основы фенилкетонурии у корейцев». Журнал генетики человека. 49 (1): 617–621. Дои:10.1007 / s10038-004-0197-5. PMID 15503242. S2CID 21446773.
- ^ а б c d е «ФКУ: устранение пробелов в лечении» (PDF). Получено 28 августа, 2020.
- ^ «Филиппинское общество по проблемам сирот - текущий реестр». psod.org.ph. Архивировано из оригинал на 2015-01-04.
- ^ Фенилкетонурия в eMedicine
- ^ Bickel, H .; Bachmann, C .; Beckers, R .; Брандт, штат Нью-Джерси; Clayton, B.E .; Corrado, G; и другие. (1981). «Массовый скрининг новорожденных на метаболические нарушения». Европейский журнал педиатрии. 137 (137): 133–139. Дои:10.1007 / BF00441305 (неактивно 22 декабря 2020 г.).CS1 maint: DOI неактивен по состоянию на декабрь 2020 г. (связь)
- ^ Ферак В., Сивакова Д., Сиглова З. (1987). "Slovenskí Cigáni (Rómovia) - populácia s najvyšším koeficientom inbrídingu v Európe". Братиславске Лекарске Листы. 87 (2): 168–175.
- ^ Борг С., Мондот С., Местре М., Каверо I. (ноябрь 1991 г.). «Никорандил: дифференциальный вклад открытия K + -каналов и стимуляции гуанилатциклазы в его сосудорасширяющее действие на различные артериальные препараты, сокращенные эндотелином-1. Сравнение с априкалимом (RP 52891) и нитроглицерином». Журнал фармакологии и экспериментальной терапии. 259 (2): 526–34. PMID 1682478.
- ^ "НПКУА> Образование> О ФКУ". npkua.org. В архиве из оригинала от 01.01.2015.
- ^ Фёллинг А. (1 января 1934 г.). "Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität". Hoppe-Seyler's Zeitschrift für Physiologische Chemie. 227 (1–4): 169–181. Дои:10.1515 / bchm2.1934.227.1-4.169.
- ^ Centerwall SA, Centerwall WR (2000). «Открытие фенилкетонурии: история молодой пары, двух больных детей и ученого». Педиатрия. 105 (1 Пет 1): 89–103. Дои:10.1542 / пед.105.1.89. PMID 10617710. S2CID 35922780.
- ^ а б Марелин Райнер-Кэнхэм, Джефф Райнер-Кэнхэм (2008), «Эвелин Хикманс», Химия была их жизнью: первые британские женщины-химики, 1880–1949, World Scientific, стр. 198, ISBN 9781908978998
- ^ Митчелл Дж. Дж., Тракадис Ю. Дж., Скрайвер К. Р. (2011). «Дефицит фенилаланингидроксилазы». Генетика в медицине. 13 (8): 697–707. Дои:10.1097 / GIM.0b013e3182141b48. PMID 21555948. S2CID 25921607.
- ^ Кох Дж (1997). Роберт Гатри - история фенилкетонурии: крестовый поход против умственной отсталости. Пасадена, Калифорния: Hope Pub. Жилой дом. С. 65–66. ISBN 0932727913. OCLC 36352725.
- ^ Kasper DC, Ratschmann R, Metz TF, Mechtler TP, Möslinger D, Konstantopoulou V, Item CB, Pollak A, Herkner KR (2010). «Национальная австрийская программа скрининга новорожденных - восьмилетний опыт работы с масс-спектрометрией. Прошлые, настоящие и будущие цели». Wiener Klinische Wochenschrift. 122 (21–22): 607–613. Дои:10.1007 / s00508-010-1457-3. PMID 20938748. S2CID 27643449.
- ^ Комровер Г.М., Сардхарвалла И.Б., Фаулер Б., Бридж С. (1979). «Региональная программа скрининга в Манчестере: 10-летняя практика ухода за пациентами и их членами». Британский медицинский журнал. 2 (6191): 635–638. Дои:10.1136 / bmj.2.6191.635. ЧВК 1596331. PMID 497752.
- ^ van Wegberg AM, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, Burlina A, Campistol J, Feillet F, Giżewska M, Huijbregts SC, Kearney S, Leuzzi V, Maillot F, Muntau AC, van Rijn М, Трефз Ф., Вальтер Дж. Х., ван Спронсен Ф. Дж. (Октябрь 2017 г.). «Полное европейское руководство по фенилкетонурии: диагностика и лечение». Журнал редких заболеваний Orphanet. 12 (1): 162. Дои:10.1186 / s13023-017-0685-2. ЧВК 5639803. PMID 29025426.
- ^ «Консенсусный документ - Э.С.ПКУ». E.S.PKU. Получено 2018-11-23.
- ^ Хагедорн Т.С., ван Беркель П., Хаммершмидт Г., Лхотакова М., Салудес Р.П. (декабрь 2013 г.). «Требования к минимальному стандарту лечения фенилкетонурии: взгляд пациентов». Журнал редких заболеваний Orphanet. 8 (1): 191. Дои:10.1186/1750-1172-8-191. ЧВК 3878574. PMID 24341788.
- ^ Burgard P, Ullrich K, Ballhausen D, Hennermann JB, Hollak CE, Langeveld M, et al. (Сентябрь 2017 г.). «Проблемы с европейскими рекомендациями по фенилкетонурии». Ланцет. Диабет и эндокринология. 5 (9): 681–683. Дои:10.1016 / S2213-8587 (17) 30201-2. PMID 28842158.
- ^ «Фенилкетонурия». Словарь Merriam-Webster.
- ^ «Фенилкетонурия». Оксфордские словари Британский словарь. Oxford University Press. Получено 2016-01-20.
- ^ «BioMarin: трубопровод: Обзор трубопровода: BMN 165 для PKU». bmrn.com. Архивировано из оригинал на 01.01.2015.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |