Синдром Сэтре-Чотцена - Saethre–Chotzen syndrome
Эта статья нужны дополнительные цитаты для проверка. (Август 2010 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
| Синдром Сэтре-Чотцена | |
|---|---|
| Другие имена |
|
 | |
| Это заболевание передается по аутосомно-доминантному типу. | |
| Специальность | Ревматология |
Синдром Сэтре-Чотцена (SCS), также известный как акроцефалосиндактилия III типа, это редкий врожденное заболевание связана с краниосиностоз (преждевременное ушивание одного или нескольких швов между костями череп ). Это влияет на форму головы и лица, в результате получается голова конуса и асимметричное лицо. У людей с SCS также есть опущенные веки (птоз ), широко расставленные глаза (гипертелоризм ), и незначительные аномалии кистей и стоп (синдактилия ).[2] Лица с более тяжелыми случаями СКС могут иметь умственную отсталость от легкой до средней степени или неспособность к обучению. В зависимости от степени тяжести, некоторым пациентам с СКС может потребоваться медицинское или хирургическое вмешательство в той или иной форме.[3] Большинство людей с СКС живут вполне нормальной жизнью, независимо от того, требуется ли им лечение или нет.[2]
Признаки и симптомы
SCS представлен в разнообразной форме. Большинство людей с СКС поражены умеренно, с неровными чертами лица и относительно плоским лицом из-за недоразвитых глазниц, скул и нижней челюсти. Помимо физических отклонений, люди с СКС также испытывают задержку роста, что приводит к относительно низкому росту. Хотя большинство людей с СКС имеют нормальный интеллект, у некоторых людей может быть легкое или умеренное умственные задержки. Более тяжелые случаи SCS с более серьезными деформациями лица возникают, когда несколько черепных швов закрываются преждевременно.[2]
Черепные дефекты

- Плоская асимметричная голова и лицо[3]
- Голова обычно конусообразная (акроцефалия ) или плоский (брахицефалия ), но также может быть длинным и узким (долихоцефалия )[4]
- Голова короткая спереди назад[5]
- Кривое лицо[4]
- Низко посаженная линия роста волос, из-за которой лоб кажется высоким и широким[5]
Дефекты рук и ног
- Лента (синдактилия ) между вторым и третьим пальцами и между вторым и третьим пальцами ноги[2]
- Короткие пальцы рук и ног (брахидактилия )[4]
- Широкий большой палец и / или широкий Hallux (большой палец) с вальгусная деформация (изгиб наружу дистальный сегмент кости / сустава)[6]
- На руках есть единственная складка для сгибания ладони[3]
Глазные дефекты
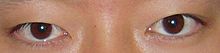

- Неравномерно расположенные глаза, которые можно пересечь (косоглазие ) или широко расставленные (гипертелоризм )[5]
- Проблемы со зрением из-за аномальной анатомии лица, которая вызывает механические нарушения экстраокулярные мышцы, что приводит к косоглазию (косоглазие)[3]
- Слезный канал стеноз (сужение слезного протока)[3]
- Опущенные веки (птоз )[3]
- Наклон вниз глазные щели (разделение между верхними и нижними веками)[3]
- Близорукость (миопия )[4]
- Эпикантальные складки (кожные складки верхнего века, закрывающие внутренний уголок глаза)[6]
- Блефарофимоз (двусторонний птоз с уменьшением размера века)[6]
- Атрофия зрительного нерва[6]
- Огнеупорные ошибки[6]
Дефекты уха, носа и рта
- Маленькие, низко посаженные уши, которые могут быть немного повернуты назад и имеют выступающие (выпуклые) уши. ушная раковина[4]
- Клювовый нос (слегка изогнутый вниз на кончике), слегка смещенный от центра и содержащий искривленная перегородка[2]
- Неправильный прикус связанные с зубными аномалиями, включая гипоплазия эмали (тонкая эмаль из-за неполного образования), гипердонтия (лишние зубы) и колышки (маленькие зубы неправильной формы)[6]
- Волчья пасть с высокой аркой[6]
Менее распространенные дефекты
- Невысокий рост[7]
- Позвоночные сращения[7]
- Врожденные проблемы с сердцем[6]
- Проблемы с речью[8]
- Анальная атрезия (уродливый прямая кишка )[3]
- Неопущенный яички (крипторхизм )[4]
- Почечные (почечные) аномалии[4]
- Расстройства личности[6]
Причины
Краниосиностоз

Череп состоит из трех основных частей, включая основание черепа (затылочная кость ), лицо (лобная кость ), а верхний (теменные кости ) и по бокам (височная кость ) головы. Большинство костей черепа постоянно остаются на месте до рождения. Однако височная и теменная кости разделены швы, которые остаются открытыми, позволяя голове немного изменять форму во время родов. Черепные швы в конечном итоге закрываются в течение первых двух лет после рождения, после того как мозг закончил рост.[2]
У пациентов с СКС коронарный шов, отделяющий лобные кости от теменных костей, закрывается преждевременно (краниосиностоз ), иногда даже до рождения. Если коронарный шов сшивается асимметрично или односторонне, то лицо и лоб будут формироваться неравномерно, из стороны в сторону. Люди с СКС имеют заостренную башнеобразную голову, потому что их мозг растет быстрее, чем их череп, что приводит к повышению внутричерепного давления (ВЧД) и вызывает выпирание макушки головы и / или лба, что способствует росту мозга. Лицо выглядит неровным, особенно в области глаз и щек, а лоб кажется широким и высоким.[2]
Из-за ненормального лба остается меньше места для развития нормальных черт лица. Это приводит к неглубоким глазницам и плоским скулам. Неглубокие глазницы делают глаза более выпуклыми или выпуклыми и вызывают большее расхождение глаз, чем обычно (гипертелоризм). Недоразвитые глазницы, скулы и нижняя челюсть делают лицо плоским. Кроме того, незначительный наклон глаз вниз вместе с опущенными веками (птоз ) добавляет к общей неровности лица.[2]
Генетика

SCS обычно наследуется как аутосомно-доминантный черта. Однако иногда дети с микроделеция 7p21 (хромосома, содержащая локус ответственные за SCS) развиваются новые аномалии и обычно демонстрируют значительные неврологические аномалии. Увеличение возраста родителей может сыграть роль в развитии новых мутаций и аномалий.[3]
Анализ связей и хромосомная перестройка выявили, что причиной SCS являются мутации в КРУТИТЬ ген (ген фактора транскрипции twist), расположенный на хромосоме 7p21. Ген TWIST кодирует основная спираль-петля-спираль (b-HLH) фактор транскрипции что контролирует голову мезенхима развитие по мере формирования черепной трубы. У людей с СКС было идентифицировано более 35 различных мутаций TWIST с участием домена b-HLH этого белка. Мутации включают промах, ерунда, и сдвиг рамки удаление /вставка мутации, которые либо укорачивают, либо разрушают домен b-HLH. У большинства людей с SCS есть одна большая делеция в области 7p21, которая содержит область, кодирующую ген TWIST.[6]
В поисках гена, ответственного за SCS, ученые Детского центра Джонса Хопкинса начали изучать ген TWIST, поскольку он влияет на мышей. Ген TWIST у мышей участвует в развитии мышц и скелета лица, головы, рук и ног. Мыши, у которых отсутствовали обе копии гена TWIST, были спонтанно абортированы до рождения и имели серьезные деформации, включая аномальные дефекты конечностей и головы и отказ нервная трубка правильно закрыть. Однако мыши с единственной копией неработающего гена TWIST выжили. Дальнейшее обследование показало, что у этих мышей были лишь незначительные дефекты черепа, кистей и стоп, подобные тем, которые наблюдаются в SCS. Ген TWIST у мышей расположен на хромосоме 12, что соответствует короткому плечу хромосома 7 в людях. Обладая этой информацией, ученые начали изолировать и карта ген TWIST человека на коротком плече хромосомы 7. Они показали, что ген TWIST человека находится в той же области, которая отсутствует у людей с SCS. При поиске различных мутаций в гене TWIST у человека было обнаружено пять различных типов мутаций у людей с SCS. Поскольку ни одна из этих мутаций не наблюдалась у нормальных людей, у которых не было SCS, это дало достаточно доказательств, чтобы сделать вывод, что ген TWIST был причинным агентом SCS1. Исследователи также изучили ген TWIST в Дрозофила (плодовая муха), чтобы определить его функцию. Они обнаружили, что в присутствии двух соединенных вместе белковых молекул TWIST ген TWIST функционирует как фактор транскрипции ДНК, то есть связывается с Двойная спираль ДНК в определенных местах, чтобы контролировать, какие гены «включаются» или активируются. Большинство идентифицированных мутаций в гене TWIST влияют на то, как белок прикрепляется к ДНК, предотвращая активацию других генов, которые обычно включаются во время развития плода.[2]
Диагностика
Пренатальная диагностика
Пренатальная диагностика Синдрома Сэтре-Чотцена при беременностях с высоким риском возможно выполнимо, но очень редко и редко выполняется. Более того, это возможно только в том случае, если мутация причина заболевания уже выявлена в семье геном. Существует несколько различных методов пренатального тестирования. Пренатальное тестирование обычно проводится примерно через 15–18 недель с использованием амниоцентез извлекать ДНК из клеток плода. Пренатальное тестирование также можно проводить в течение 10–12 недель с использованием отбор проб ворсинок хориона (CVS) для извлечения ДНК из плода.[7] В последнее время наблюдается повышенный интерес к использованию УЗИ оборудование для выявления аномалий черепа плода из-за незрелого сращения черепные швы.[4]
Клинический диагноз
Общий диагноз SCS в первую очередь основан на клинических данных и наблюдениях, основанных на исследовании дисморфологии (оценка структурных дефектов) и рентгенологической оценке (Рентгеновские лучи, МРТ, и Компьютерная томография ).[6]
Молекулярная / генетическая диагностика
Клинический диагноз SCS может быть подтвержден путем тестирования TWIST1 ген (только ген в котором мутации как известно, вызывают SCS) для мутаций с использованием анализа ДНК, например анализ последовательности, удаление /дублирование анализ и цитогенетика / РЫБЫ анализ. Анализ последовательности экзон 1 (TWIST1 кодирующая область ) представляет собой хороший метод определения частоты мутаций в гене TWIST1. Эти мутации включают ерунда, промах, мутация сайта сплайсинга, и внутригенные удаления /вставки. Анализ делеции / дупликации выявляет мутации в гене TWIST1, которые нелегко обнаружить с помощью анализа последовательности. Общие методы включают ПЦР, мультиплексное лигирование-зависимое усиление зонда (MLPA), и хромосомный микрочип (ХМА). Цитогенетический / FISH-анализ прикрепляет флуоресцентные метки ДНК-маркеры к денатурированной хромосома и затем исследуется при флуоресцентном освещении, которое выявляет мутации, вызванные транслокации или же инверсии с участием 7p21. Иногда у людей с SCS наблюдается транслокация хромосом, инверсия или кольцевая хромосома 7 с участием 7p21, что приводит к атипичным находкам, таким как увеличенная задержка развития.[7] Люди с СКС обычно имеют нормальное функционирование мозга и редко имеют психические нарушения. По этой причине, если у человека есть как SCS, так и умственная отсталость, то им следует более тщательно проверить свой ген TWIST1, потому что это не является нормальным признаком SCS.[2] Цитогенетическое тестирование и прямое генное тестирование также может использоваться для изучения дефектов генов / хромосом. Цитогенетическое тестирование - это исследование хромосом для обнаружения увеличения или уменьшения хромосом или сегментов хромосом с использованием флуоресцентной гибридизации in situ (FISH) и / или сравнительная геномная гибридизация (CGH). В прямом генном тестировании используются кровь, волосы, кожа, амниотическая жидкость, или другие ткани, чтобы найти генетические нарушения. Прямое генное тестирование может определить, есть ли у человека SCS, путем тестирования крови человека на мутации в гене TWIST1.[7]
Дифференциальная диагностика
Генетическое тестирование позволяет поставить окончательный диагноз, потому что оно позволяет дифференцировать сходные состояния друг от друга на основе того, какой ген мутировал.[2] В следующей таблице приведены условия, аналогичные SCS:
| Условие | Симптомы | Ген |
|---|---|---|
| SCS | Широко расставленные глаза, низкая линия волос, опущенные глаза, межпальцевые перепонки, деформированные уши, косоглазие и наклон вниз глазные щели | TWIST1 |
| Синдром Робиноу-Сорауфа | Широко расставленные глаза, искривленная перегородка, плоский череп сзади, деформированные уши, скрещенные глаза, выступающая челюсть и удвоение дистальной фаланги | TWIST1 |
| Синдром Мюнке | Широко расставленные глаза, увеличенная голова, потеря слуха, плоские щеки и низко посаженные уши | FGFR3 |
| Синдром Крузона | Широко расставленные глаза, коротко-широкая голова, потеря слуха, выпученные глаза, клювый нос, низко посаженные уши, косоглазие, выступающий подбородок и короткие плечевая и бедренная кость | FGFR2 & FGFR3 |
| Синдром Пфайффера | Широко расставленные глаза, недоразвитая челюсть, клювый нос, потеря слуха и выпученные глаза | FGFR1 & FGFR2 |
| Синдром Аперта | Широко расставленные глаза, выступающий лоб, плоская задняя часть черепа, выпученные глаза, низко посаженные уши, плоское или вогнутое лицо, короткий большой палец и перепончатые пальцы | FGFR2 |
| Изолированный односторонний коронарный синостоз | Единственный порок развития - это преждевременное срастание швов; Если не лечить, может привести к асимметрии лица, напоминающей СКС. | FGFR (любой) |
| Синдром Баллера-Герольда (BGS) | Короткая широкая голова, выпученные глаза, плоский лоб, пойкилодермия, лучевая деформация с уменьшенным количеством пальцев, недоразвитие или отсутствие большого пальца и лучевой кости, а также задержка роста | RECQL4 |
Уход


Физические отклонения, возникающие в результате SCS, обычно легкие и требуют лишь незначительной хирургической процедуры или вообще ее отсутствия. Одним из распространенных симптомов СКС является развитие коротких (брахидактилия ), перепончатые пальцы рук и широкие пальцы ног (синдактилия ). Эти характеристики не вызывают каких-либо проблем с функционированием рук или ног, и, следовательно, не требуется никаких медицинских процедур для исправления аномалий, если только пациент этого не попросит. Переплетение пальцев может повлиять на основание пальцев, что приведет к задержке роста кисти в детстве, но не вызывает функциональных нарушений. Иногда у людей с СКС развиваются широкие пальцы ног, потому что кости на концах пальцев ног дублируются. Это особенно заметно на большом пальце ноги, но не требует хирургического вмешательства, поскольку не влияет отрицательно на общую функцию стопы. Люди с такими аномалиями пальцев ног ходят нормально и могут носить обычную обувь.[9]
В более тяжелых случаях требуются частые операции и клинический контроль на протяжении всего развития. Ребенок родился с асимметричным односторонним венечным синостоз должен пройти краниопластика в течение первого года жизни, чтобы предотвратить увеличение внутричерепное давление и предотвратить прогрессирующее асимметрия лица. Краниопластика - это хирургическая процедура по исправлению преждевременного сращения костей черепа. Операция направлена на реконструкцию и репозицию костей и швов для обеспечения наиболее нормального роста.[6] Краниопластика необходима для продолжения роста и важна по двум основным причинам. Прежде всего, череп должен иметь возможность приспособиться к растущему мозгу после родов, чего он не может, потому что череп не растет так быстро, как мозг, пока швы остаются слитными. Это приводит к увеличению давления, окружающего мозг, и препятствует росту мозга, в результате чего у человека возникают серьезные проблемы, которые, если его не лечить, могут в конечном итоге привести к смерти. Во-вторых, краниопластика может потребоваться по внешнему виду.[7] Это особенно характерно для людей с асимметричным односторонним коронарным синостозом, который требует реконструктивная хирургия лица и черепа. Если краниопластика не выполняется, особенно у людей с односторонним коронарным синостозом, асимметрия лица со временем будет ухудшаться, поэтому краниопластику следует проводить как можно скорее.[9]
Хирургия также может потребоваться людям с проблемами зрения. Проблемы со зрением обычно возникают из-за недостатка места в глазной орбите и черепе из-за аномальной костной структуры лица. Уменьшение пространства также может привести к неправильному или отсутствию слезные протоки и повреждение нервов. Реконструктивная хирургия обычно требуется для увеличения краниального пространства, исправления слезного протока. стеноз, и / или правильно птоз век, чтобы предотвратить амблиопия (ленивый глаз).[2]
Операция на средней части лица также может потребоваться в раннем детстве для устранения респираторных заболеваний, неправильный прикус, и трудности с глотанием. А волчья пасть также корректируется хирургическим путем и может включать использование тимпаностомические трубки. При необходимости индивидуум пройдет ортогнатическое лечение и / или ортодонтическое лечение после завершения развития лица.[2] Поскольку потеря слуха часто связана с СКС, рекомендуется проводить аудиологический скрининг на протяжении всего детства.[6]
После черепно-реконструктивной хирургии ребенку может потребоваться носить формовочный шлем или какой-либо другой вид защиты головы, пока кости черепа не встанут на место. Обычно это занимает около трех месяцев и зависит от возраста ребенка и тяжести состояния. После выздоровления люди с СКС выглядят и ведут себя совершенно нормально, поэтому никто даже не сможет сказать, что у них СКС.[10]
Эпидемиология
СКС - самый распространенный краниосиностоз синдром и поражает 1 из 25 000–50 000 человек.[11] Он встречается у всех расовых и этнических групп и в равной степени поражает мужчин и женщин.[2] Если родитель несет копию мутации гена SCS, то есть 50% -ная вероятность, что их ребенок также будет нести копию мутации гена, и в этом случае ребенок может проявлять или не проявлять признаки SCS. Также существует 50% -ная вероятность, что у их ребенка будут две рабочие копии гена, и, следовательно, у него не будет SCS. Если оба родителя несут одну копию мутации гена SCS, то существует 25% вероятность того, что у их ребенка будут две копии мутации гена (так что у ребенка разовьется тяжелая форма SCS), вероятность 25%, что у их ребенка будут две нормальные копии ген (это было бы совершенно нормально), и с вероятностью 50% их ребенок будет нести одну копию мутации гена и 1 нормальную копию (так что ребенок может отображать или не отображать SCS).[2] В редких случаях у двух нормальных родителей может быть ребенок с SCS из-за de novo мутация. Точная причина de novo мутация неизвестна, но, похоже, она не связана ни с чем, что родители делали или не делали во время беременности.[12] СКС из-за de novo мутации настолько редки, что доля прошлых случаев неизвестна.[7]
История
В 1931 году норвежец Хокон Сэтре психиатр, описал схожие характеристики между матерью и двумя ее дочерьми. У всех были длинные и неровные черты лица, низко посаженные волосы, короткие пальцы и перепонка между вторым и третьим пальцами, а также между вторым, третьим и четвертым пальцами ног. Годом позже, в 1932 г., немец Ф. Чотцен психиатр, описал отца и двух его сыновей как имеющих очень похожие характеристики, как мать и ее дочери, а также как имеющих потерю слуха, низкий рост и умеренную умственную отсталость. Следовательно, название «Синдром Саэтре-Чотцена» было получено от двух ученых, которые отдельно описали это состояние без каких-либо предварительных сведений о другом.[2]
Рекомендации
- ^ "Здоровье детей: синдром Сэтре Чотцен". WebMD. Получено 28 ноября, 2012.
- ^ а б c d е ж грамм час я j k л м п о п Бланчфорд, Стейси L (2002). Энциклопедия генетических заболеваний Гейла. Мичиган: Гейл Групп. С. 1019–1021. ISBN 9780787656140.
- ^ а б c d е ж грамм час я Аллансон, Джудит, Кэссиди, Сюзанна (2010). Управление генетическими синдромами. Нью-Джерси: John Wiley & Sons, Inc., стр. 230–235. ISBN 9780470191415.
- ^ а б c d е ж грамм час Винбрандт, Джеймс (2008). Генетические заболевания и врожденные пороки. Нью-Йорк: Факты о File, Inc., стр.340. ISBN 9780816063963.
- ^ а б c «Синдром Сэтре-Чотцена». Международный черепно-лицевой институт. Получено 28 октября, 2012.
- ^ а б c d е ж грамм час я j k л м Клаузер Л., Гали М. «Синдром Сэтре-Чотцена» (PDF). Orphanet. Получено 28 октября, 2012.
- ^ а б c d е ж грамм Галлахер Э., Ратизунторн С., Каннингем М. (1993). «Синдром Сэтре-Чотцена». Синдром Сэтре Чотцен. NCBI. PMID 20301368. Получено 28 октября, 2012.
- ^ «Синдром Сэтре-Чотцена». Детская больница и медицинский центр. Получено 25 октября, 2012.
- ^ а б Андерсон, Питер. «Заголовки черепно-лицевой поддержки» (PDF). Архивировано из оригинал (PDF) 30 июня 2012 г.. Получено 27 ноября, 2012.
- ^ «Хирургические варианты краниосиностоза». Johns Hopkins Medicine. Получено 28 ноября, 2012.
- ^ «Синдром Сэтре-Чотцена». Бостонская детская больница. Получено 28 ноября, 2012.
- ^ «Синдром Сэтре-Чотцена». Детская больница Сиэтла. Получено 28 ноября, 2012.
внешняя ссылка
- Резюме NIH по SCS
- НАЦИОНАЛЬНЫЕ ИНСТИТУТЫ ЗДРАВООХРАНЕНИЯ США. Синдромы множественной врожденной аномалии / умственной отсталости (MCA / MR)
- Статья GeneReview / NCBI / NIH / UW о синдроме Сэтре – Чотцена
| Классификация | |
|---|---|
| Внешние ресурсы |