Синдром аперта - Apert syndrome
| Синдром аперта | |
|---|---|
| Другие имена | Акроцефало-синдактилия 1 типа[1] |
 | |
| Синдром Хэнда Аперта с синдактилия | |
| Специальность | Медицинская генетика |
Синдром аперта это форма акроцефалосиндактилия, а врожденное заболевание характеризуется пороками развития черепа, лица, кистей и стоп. Классифицируется как синдром жаберной дуги, поражающий первую жаберная (или глоточная) дуга, предшественник верхняя челюсть и нижняя челюсть. Нарушения развития жаберных дуг в процессе развития плода оказывают стойкое и широко распространенное воздействие.
В 1906 г. Эжен Аперт Французский врач описал девять человек, обладающих схожими атрибутами и характеристиками.[2] Лингвистически в термине «акроцефалосиндактилия» акро является Греческий для «пика» относится к «остроконечной» голове, которая часто встречается при синдроме; головной мозг, также из греческого, это сочетание формы означает «голова»; синдактилия относится к перепонкам пальцев рук и ног.
В эмбриология, руки и ноги имеют селективные клетки, которые умирают в процессе, называемом селективной смертью клеток, или апоптоз, вызывая разделение цифр. В случае акроцефалосиндактилии избирательной гибели клеток не происходит, и кожа, а в редких случаях кости между пальцами рук и ног срастается.
Также поражаются кости черепа, аналогично Синдром Крузона и Синдром пфайффера. Краниосиностоз возникает, когда череп плода и лицевые кости срастаются слишком рано в утробе, нарушая нормальный рост костей. Слияние разных швы приводит к разным образцам роста черепа. Примеры включают: тригоноцефалия (слияние метопический шов ), брахицефалия (слияние коронарный шов и ламбдовидный шов двусторонне), долихоцефалия (слияние сагиттальный шов ), плагиоцефалия (слияние коронарного и ламбдовидные швы в одностороннем порядке) и оксицефалия или туррицефалия (слияние коронарных и лямбдовидных швов).
Данные о заболеваемости синдромом среди населения различаются.[3] по оценкам, только 1 рождение из 200 000[4] и 160 000 даны в среднем по старым исследованиям.[5][6] Однако исследование, проведенное в 1997 году Калифорнийской программой мониторинга врожденных дефектов, показало, что уровень заболеваемости составляет 1 на 80 645 из почти 2,5 миллиона живорождений.[7] Другое исследование, проведенное в 2002 году Черепно-лицевым центром Детской больницы Северного Техаса, показало, что заболеваемость выше - примерно 1 на 65 000 живорождений.[3]
Признаки и симптомы
Краниосиностоз
Черепные аномалии - наиболее очевидные последствия акроцефалосиндактилии. Краниосиностоз происходит, при котором черепные швы закрываются слишком рано, хотя мозг ребенка все еще растет и расширяется. Брахицефалия это обычная модель роста, где коронковые швы закрываться преждевременно, не позволяя черепу расширяться вперед или назад и заставляя мозг расширять череп в стороны и вверх. Это приводит к еще одной общей характеристике - высокому выступающему лбу с плоской задней частью черепа. Из-за преждевременного закрытия коронарных швов может развиться повышенное черепное давление, что приводит к умственной отсталости. Плоское или вогнутое лицо может развиться в результате недостаточного роста срединных костей лица, что приводит к состоянию, известному как псевдомандибулярный прогнатизм. Другие особенности акроцефалосиндактилии могут включать неглубокие костные орбиты и широко расставленные глаза. Низко посаженные уши также являются типичной характеристикой синдромов жаберных дуг.
Синдактилия
Все синдромы акроцефалосиндактилии демонстрируют определенный уровень аномалий конечностей, поэтому их бывает трудно отличить друг от друга. Однако типичные деформации кисти у пациентов с синдромом Аперта отличают его от других синдромов.[8] Руки у пациентов с синдромом Аперта всегда имеют четыре общие черты:[9]
- короткий большой палец с радиальным отклонением
- сложный синдактилия указательного, длинного и безымянного пальца
- симбрахифалангизм
- простая синдактилия четвертого веб-пространства
Деформация пространства между указательным и большим пальцами может быть переменной. Основываясь на этом первом веб-пространстве, мы можем различить три различных типа деформации рук:
- Тип I: Также называется «лопатой». Самый частый и наименее тяжелый вид деформации. Большой палец показывает радиальное отклонение и клинодактилия но отделен от указательного пальца. Указательный, длинный и безымянный пальцы срастаются в дистальных межфаланговых суставах и образуют плоскую ладонь. Во время эмбриональной стадии слияние не влияет на продольный рост этих пальцев, поэтому они имеют нормальную длину. В четвертом веб-пространстве мы всегда видим простую синдактилию, полную или неполную.
- Тип II: Также называется «ложкой» или «рукавицей». Это более серьезная аномалия, поскольку большой палец срастается с указательным пальцем простой полной или неполной синдактилией. Только дистальная фаланга большого пальца не соединяется в костном сращении с указательным пальцем и имеет отдельный ноготь. Поскольку сращение пальцев происходит на уровне дистальных межфаланговых суставов, образуется вогнутая ладонь. В большинстве случаев мы видим полную синдактилию четвертого веб-пространства.
- Тип III: Также называется рукой «копыто» или «бутон розы». Это самая редкая, но также и самая тяжелая форма деформации руки при синдроме Аперта. Это твердое сращение всех пальцев костей и хрящей с одним длинным соединенным ногтем. Большой палец повернут внутрь, и часто невозможно различить пальцы. Обычно получить правильное изображение руки очень сложно из-за перекрытия костей, но одного физического осмотра недостаточно для определения степени деформации.
| Тип I ("лопата") | Тип II ("рукавица") | Тип III ("бутон розы") | |
|---|---|---|---|
| Первое веб-пространство | Простая синдактилия | Простая синдактилия | Сложная синдактилия |
| Средние три пальца | Поперечное слияние с плоской ладонью | Слияние кончиков пальцев, образующих вогнутую ладонь | Плотное сращение всех цифр одним сросшимся ногтем |
| Четвертое веб-пространство | Простая и неполная синдактилия | Простая и полная синдактилия | Простая и полная синдактилия |
Стоматологическое значение
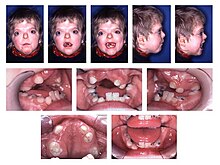
Общие характерные черты акроцефалосиндактилии - высокое арочное небо, псевдомандибулярный прогнатизм (появляется как нижнечелюстной прогнатизм ), узкое небо и скученность зубов.
Другие признаки
Омфалоцеле был описан у двух пациентов с синдромом Аперта Германом Т.Е. и другие. (США, 2010) и Ercoli G. et al. (Аргентина, 2014 г.). Омфалоцеле - это врожденный дефект, при котором кишечник или другие органы брюшной полости находятся вне тела младенца из-за отверстия в области пупка. Однако связь между омфалоцеле и синдромом Аперта еще не подтверждена, поэтому необходимы дополнительные исследования.[10][11]
Причины
Акроцефалосиндактилия может быть аутосомно-доминантный беспорядок. Мужчины и женщины страдают одинаково; однако исследования еще не установили точную причину. Тем не менее, почти все случаи носят спорадический характер, что свидетельствует о новых мутациях или экологическом оскорблении. геном. Потомство родителей с синдромом Аперта имеет 50% шанс унаследовать это заболевание. В 1995 году A.O.M. Уилки опубликовал статью, в которой представлены доказательства того, что акроцефалосиндактилия вызвана дефектом рецептор фактора роста фибробластов 2 ген, на хромосома 10.[12]
Синдром Аперта - аутосомно-доминантное заболевание; примерно две трети случаев связаны с мутацией C в G в позиции 755 в FGFR2 ген, который вызывает изменение Ser в Trp в белке.[13] Это горячая точка мужской мутации: в исследовании 57 случаев мутация всегда происходила на аллеле, полученном от отца.[14] Исходя из наблюдаемой распространенности заболевания при рождении (1 из 70000), очевидная частота мутаций C в G на этом участке составляет около 0,00005, что в 200-800 раз выше, чем обычная частота мутаций в CG. динуклеотиды. Причем заболеваемость резко возрастает с увеличением возраст отца. Goriely et al. (2003) проанализировали аллельное распределение мутаций в образцах спермы мужчин разного возраста и пришли к выводу, что простейшим объяснением данных является то, что мутация C в G дает клетке преимущество в мужской зародышевой линии.[13]
До сих пор не очень ясно, почему у людей с синдромом Аперта наблюдается краниосиностоз и синдактилия. Одно исследование предполагает, что это как-то связано с выражением трех изоформы FGFR2, гена с точечные мутации что вызывает синдром у 98% пациентов.[15] KGFR, рецептор фактора роста кератиноцитов, представляет собой изоформу, активную в метафиз и межфаланговые суставы. FGFR1 - изоформа, активная в диафиз. FGFR2-Bek активен в метафизе, а также в диафизе, но также и в межпальцевом суставе. мезенхима. Точечная мутация увеличивает лиганд-зависимую активацию FGFR2 и, следовательно, его изоформ. Это означает, что FGFR2 теряет свою специфичность, вызывая связывание FGF, которые обычно не связываются с рецептором.[16] Поскольку FGF подавляет апоптоз, межпальцевая мезенхима сохраняется. FGF также увеличивает репликацию и дифференциацию остеобласты, отсюда раннее сращение нескольких швов черепа. Это может объяснить, почему при синдроме Аперта всегда обнаруживаются оба симптома.[нужна цитата ]
Диагностика
Диагноз обычно ставится по видимым физическим характеристикам и может быть подтвержден черепом. рентгеновский снимок или же КТ-исследование головы. Молекулярный генетическое тестирование может подтвердить диагноз.[17]
Лечение
Краниосиностоз
Операция необходима, чтобы наложение коронарных швов не повредило развитию мозга. В частности, операции на ЛеФорт III или моноблочный мидфейс дистракционный остеогенез которые отделяют среднюю часть лица или всю верхнюю часть лица, соответственно, от остальной части черепа, выполняются для их перемещения в правильной плоскости. Эти операции выполняются как пластическими, так и челюстно-лицевыми хирургами, часто в сотрудничестве.[нужна цитата ]
Синдактилия
В Apert нет стандартного лечения пороков развития рук из-за различий и тяжести клинических проявлений у разных пациентов. Поэтому к каждому пациенту следует подходить и лечить индивидуально, стремясь к адекватному балансу между функциональностью руки и эстетикой. Однако в зависимости от степени деформации можно дать некоторые рекомендации. В общем, изначально рекомендуется освободить первое и четвертое межпальцевые промежутки, тем самым освободив пограничные лучи.[18] Это позволяет ребенку брать предметы руками, что является очень важной функцией для его развития. Позже необходимо освободить второе и третье межпальцевые промежутки. Поскольку в Apert есть три типа рук, все со своими деформациями, всем им нужен другой подход к их лечению:[нужна цитата ]
- Рука типа I обычно нуждается только в межпальцевом выпуске веб-пространства. Первый веб-релиз нужен редко, но часто необходимо его углубление. Потребуется коррекция клинодактилии большого пальца.
- В руках типа II рекомендуется вначале высвободить первый и пятый лучи, затем необходимо освободить второе и третье межпальцевые промежутки паутины. Также необходимо исправить клинодактилию большого пальца. Может потребоваться удлинение фаланги большого пальца, таким образом увеличивая первое пространство паутины. Как при типе I, так и при типе II рецидивирующая синдактилия второго веб-пространства будет возникать из-за псевдоэпифиза в основании указательной пястной кости. Это должно быть исправлено более поздними версиями.
- Руки типа III лечить сложнее всего из-за их сложности. Прежде всего, рекомендуется освободить первое и четвертое веб-пространство, тем самым преобразовав его в руку типа I. Лечение мацерации и инфекций ногтевого ложа также следует проводить вначале. Для увеличения первого интервала можно удлинить большой палец. Предполагается, что в тяжелых случаях следует рассмотреть возможность ампутации указательного пальца. Однако, прежде чем принять это решение, важно взвесить возможное улучшение, которое должно быть достигнуто, с возможными психологическими проблемами ребенка позже из-за эстетики руки. Позже следует освободить второе и / или третье межпальцевое веб-пространство.
По мере роста ребенка и соответственно рук необходимы вторичные ревизии для лечения контрактур и улучшения эстетики.
Смотрите также
Рекомендации
- ^ «Синдром Аперта | Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS». rarediseases.info.nih.gov. Получено 18 мая 2019.
- ^ синд / 194 в Кто это назвал?
- ^ а б Фирон, Джеффри А. (июль 2003 г.). «Лечение рук и ног при синдроме Аперта: эволюция в управлении». Пластическая и реконструктивная хирургия. Даллас, Техас. 112 (1): 1–12. Дои:10.1097 / 01.PRS.0000065908.60382.17. ISSN 0032-1052. PMID 12832871. S2CID 8592940.
- ^ Форман, Фил (2009). Обучение студентов с умственной отсталостью: исследования и практика (PB). ИАП. п. 30. ISBN 978-1-60752-214-0.
- ^ Картер, Чарльз Х. (1965). Медицинские аспекты умственной отсталости. Томас. п. 358. OCLC 174056103.
- ^ Эйб Берт Бейкер; Лоуэлл Х. Бейкер (1979). «Синдром Аперта». Клиническая неврология. Медицинский отдел, Harper & Row. 3: 47. OCLC 11620265.
- ^ Эйткен, Дж. Кеннет (2010). Генетические факторы аутизма: справочник для профессионалов. Джессика Кингсли. п. 133. ISBN 978-1-84310-976-1.
- ^ Каплан, Л. К. (апрель 1991 г.). «Клиническая оценка и многопрофильное лечение синдрома Аперта». Клиники пластической хирургии. 18 (2): 217–25. Дои:10.1016 / S0094-1298 (20) 30817-8. ISSN 0094-1298. PMID 2065483.
- ^ Аптон, Дж. (Апрель 1991 г.). «Синдром Аперта. Классификация и патологическая анатомия аномалий конечностей». Клиники пластической хирургии. 18 (2): 321–55. Дои:10.1016 / S0094-1298 (20) 30826-9. ISSN 0094-1298. PMID 2065493.
- ^ Herman, TE; Сигел, MJ (октябрь 2010 г.). «Синдром Аперта с омфалоцеле». J. Perinatol. 30 (10): 695–697. Дои:10.1038 / jp.2010.72. PMID 20877364.
- ^ Ercoli, G; Бидондо, депутат; Сенра, Британская Колумбия; Гройсман, Б. (сентябрь 2014 г.). «Синдром Аперта с омфалоцеле: история болезни». Исследование врожденных пороков, часть A: клиническая и молекулярная тератология. 100 (9): 726–729. Дои:10.1002 / bdra.23270. PMID 25045033.
- ^ Wilkie, A O; С. Ф. Слэйни; М Олдридж; М. Д. Пул; Дж. Дж. Эшворт; А. Д. Хокли; Р. Д. Хейворд; Д. Дж. Дэвид; Л. Дж. Пуллейн; П. Ратленд (февраль 1995 г.). «Синдром Аперта является результатом локализованных мутаций FGFR2 и является аллельным с синдромом Крузона». Природа Генетика. 9 (2): 165–72. Дои:10.1038 / ng0295-165. PMID 7719344. S2CID 12423131.
- ^ а б Гориели, А .; Маквин, Джорджия; Рёймир, М; Ингемарссон, Б. Уилки, АО (2003). «Доказательства избирательного преимущества патогенных мутаций FGFR2 в мужской зародышевой линии». Наука. 301 (5633): 643–6. Bibcode:2003Наука ... 301..643G. Дои:10.1126 / science.1085710. PMID 12893942. S2CID 33543066.
- ^ Молони, DM; Slaney, SF; Oldridge, M; Wall, SA; Сахлин, П; Стенман, Г; Уилки, АО (1996). «Исключительное отцовское происхождение новых мутаций при синдроме Аперта». Природа Генетика. 13 (1): 48–53. Дои:10.1038 / ng0596-48. PMID 8673103. S2CID 26465362.
- ^ Бритто, Дж. А; J C T Chan; Р. Д. Эванс; Р. Д. Хейворд; Б. М. Джонс (май 2001 г.). «Дифференциальная экспрессия рецепторов фактора роста фибробластов в цифровом развитии человека предполагает общий патогенез сложной акросиндактилии и краниосиностоза». Пластическая и реконструктивная хирургия. 107 (6): 1331–1338. Дои:10.1097/00006534-200105000-00001. PMID 11335797. S2CID 32124914.
- ^ Hajihosseini MK, Duarte R, Pegrum J, Donjacour A, Lana-Elola E, Rice DP, Sharpe J, Dickson C (февраль 2009 г.). «Доказательства того, что Fgf10 способствует скелетным и висцеральным дефектам модели мышей с синдромом Аперта». Dev. Dyn. 238 (2): 376–85. Дои:10.1002 / dvdy.21648. PMID 18773495. S2CID 39997577.
- ^ «Синдром Аперта | Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS». rarediseases.info.nih.gov. Получено 17 марта 2018.
- ^ Цукер, Р. М. (апрель 1991 г.). «Синдактильная коррекция кисти при синдроме Аперта». Клиники пластической хирургии. 18 (2): 357–64. Дои:10.1016 / S0094-1298 (20) 30827-0. ISSN 0094-1298. PMID 1648464.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |