Открытие и разработка ингибиторов фосфодиэстеразы 5 - Discovery and development of phosphodiesterase 5 inhibitors
Эта статья должна быть обновлено. (Март 2017 г.) |
Фосфодиэстеразы (PDE) являются надсемейство из ферменты. Это суперсемейство далее классифицируется на 11 семейств, PDE1 - PDE11, на основе регуляторных свойств: аминокислота последовательности, специфичность субстрата, фармакологические свойства и распределение в тканях. Их функция - разлагать внутриклеточные вторые мессенджеры такие как циклический аденинмонофосфат (лагерь ) и циклический гуанозинмонофосфат (cGMP ), что приводит к нескольким биологическим процессам, таким как влияние на уровень внутриклеточного кальция Ca2+ путь.[1]
Фосфодиэстераза 5 (PDE5 ) широко экспрессируется в нескольких тканях организма, например в головном мозге, легких, почках, мочевом пузыре, гладких мышцах и тромбоцитах.[1] Можно предотвратить гидролиз цГМФ путем ингибирования ФДЭ5 и, следовательно, лечить заболевания, связанные с низким уровнем цГМФ, из-за этого ФДЭ5 является идеальной мишенью для разработки ингибиторов.[2] Терапевтические эффекты ингибирования PDE5 были продемонстрированы в нескольких сердечно-сосудистый условия, хроническая болезнь почек и сахарный диабет.[3]
Главная Ингибиторы ФДЭ5 (подмножество ингибиторы фосфодиэстеразы ) находятся силденафил, тадалафил, варденафил, и аванафил, и хотя все они имеют один и тот же механизм действия, каждый из них имеет уникальный фармакокинетический и фармакодинамический свойства, которые определяют их пригодность в различных условиях и профиль их побочных эффектов.[3]
Общий
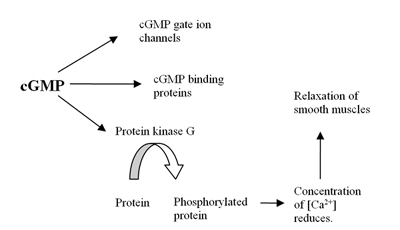
В человеческий геном содержит не менее 21 гены участвует в определении внутриклеточных уровней лагерь и cGMP выражением фосфодиэстераза белки или PDE. Эти PDE сгруппированы по крайней мере в 11 функциональных подсемейств, называемых PDE1-PDE11.[4] ФДЭ - это ферменты, которые гидролизовать циклический аденозин-3,5-монофосфат (цАМФ) и циклический гуанозин 3,5-монофосфат (цГМФ), которые являются внутриклеточными вторые мессенджеры, в AMP и GMP. Эти вторичные посланники контролируют многие физиологические процессы.[5]В лагерь сформирован из АТФ посредством фермент аденилилциклаза и cGMP сформирован из GTP ферментом гуанилилциклаза которые либо мембрана связанный или растворимый в цитозоль. Когда растворимый он функционирует как рецептор за оксид азота (НЕТ) (см. Рисунок 1).[6]Формирование cGMP инициирует несколько реакций в организме, включая влияние на цГМФ ионные каналы, cGMP привязка белки и протеинкиназа G (PKG). Воздействие на PKG снижает уровни кальций ведущий к расслаблению гладкие мышцы (см. рисунок 2).[7] Фермент PDE5 специфичен для cGMP это означает, что он только гидролизует cGMP но не цАМФ.[8] Избирательность опосредована сложной сетью водородная связь что благоприятно для цГМФ, но неблагоприятно для цАМФ в ФДЭ5.[9]За счет ингибирования фермента PDE5 cGMP концентрация будет повышена и, следовательно, может увеличить расслабление гладких мышц.[7] PDE5 имеет только один подтип, PDE5A, из которых у человека есть 4 изоформы, называемые PDE5A1-4.[8] Разница в изоформах PDE5A1-3 заключается только в 5´ конце мРНК и соответствующем N-конце белка.[10]

Распределение ФДЭ5 в организме
У людей распределение изоформ PDE5A1 и PDE5A2 такое же, и их можно найти в мозг, легкое ткань, сердце, печень, почки, мочевой пузырь, предстательная железа, уретра, пенис, матка и скелетные мышцы. PDE5A2 встречается чаще, чем PDE5A1. PDE5A3 не так широко распространен, как две другие изоформы, и встречается только в гладкая мышца тканях, он находится в сердце, мочевой пузырь, предстательная железа, уретра, пенис и матка,[10][11] Точное распределение изоформы PDE5A4 в литературе не обнаружено. Фермент PDE5 у людей также был зарегистрирован в тромбоциты, желудочно-кишечный эпителиальный клетки Клетки Пуркинье из мозжечок,[12] кавернозное тело,[5] поджелудочная железа,[13] плацента и толстая кишка,[4] кавернозное тело клитора а также гладкие мышцы и эпителий влагалища.[11]
Структура PDE и SAR
Ферменты PDE состоят из 3 функциональных доменов: N-концевой циклиновый складчатый домен, линкерный спиральный домен и C-терминал домен спирального пучка (см. рисунок 3).[9] Активный сайт представляет собой глубокий карман на стыке трех субдоменов и выстлан высококонсервативными остатками между изотипы PDE.[14] Глубина кармана составляет примерно 15 Å, а размер отверстия - примерно 20 на 10 Å. Объем активного центра составляет от 875 до 927 Å.3.[14] В активный сайт PDE5 был описан как подразделенный на 3 основных региона на основе его Кристальная структура в комплексе с силденафил:[7]
- Сайт M: содержит как цинк и магний ион. Роль ионов заключается в стабилизации структуры и активации гидроксида, чтобы опосредовать реакцию. Современные ингибиторы PDE5 не взаимодействуют с ионами металлов, в отличие от cGMP. Прямые или косвенные взаимодействия могут улучшить эффективность будущих ингибиторов.[9]
- Q-карман: считается, что гуанидиновая группа цГМФ связывается в этой области, поскольку Q-карман вмещает пиразолопиримидинон группа (см. рисунок 4) силденафил. Пиразолопиримидинон из силденафил имитирует гуанин в цГМФ и имеет те же донорные и акцепторные свойства Н-связи, образуя бидендратную Н-связь с Q817. Карта и другие. Опишите Q-карман, разделенный на 3 части:[14]
- Седло, образованное законсервированными глутамин (Q817 в PDE5A, Q443 в PDE4B и Q369 в PDE4D) и P-зажим (гидрофобный зажим на узкой стороне кармана активных центров, образованный инвариантным пурин-селективным глутамин и пара консервативных остатков).[14]
- 2 узких, гидрофобный карманы Q1 и Q2, состоящие в основном из гидрофобный остатки по бокам седла.[14]
- L-область: метилпиперазиновая группа (см. Фиг. 4) силденафил окружен остатками Tyr 664, Met 816, Ala 823 и Gly 819, а остатки 662-664 образуют крышку над карманом, сужая вход в активный сайт PDE5.
Чон и другие.[9] также описывают четвертый карман, называемый H-карманом, который является гидрофобным и вмещает этоксифенильную группу силденафил 3 ингибитора PDE5 уже присутствуют на рынке, силденафил, тадалафил и варденафил, занимают часть активного сайта, в основном вокруг Q-кармана, а иногда и M-кармана, и все 3 взаимодействуют с активным сайтом тремя важными способами:
- взаимодействие между ионами металлов через воду
- водородная связь с седлом Q-кармана
- гидрофобное взаимодействие с гидрофобными остатками, выстилающими полость активного центра.[14]
Также было описано, что гидрофобное взаимодействие с карманами Q1 и Q2 важно для эффективности ингибитора, и различия между изотипами PDE в кармане Q2 могут использоваться для селективности между изотипами.[14]


Роль в болезнях
Эректильная дисфункция
Лекарства, подавляющие ФДЭ5, силденафил, тадалафил и варденафил, использовались для лечения Эректильная дисфункция.[16] Эти ингибиторы увеличивают цГМФ, расслабление гладких мышц и, как следствие, вызывают эрекцию полового члена.[9] во время сексуальной стимуляции.[17]
Легочная артериальная гипертензия
Повышенная регуляция экспрессии гена PDE5 наблюдалась на животных моделях легочной гипертензии и, как полагают, способствует вазоконстрикция в легком.[3] Несколько рандомизированных контролируемых исследований по изучению использования ингибиторов ФДЭ5 при легочной артериальной гипертензии, подтипе легочной гипертензии, продемонстрировали их сильные эффекты в снижении легочной гипертензии и ремоделирования сосудов, а также в улучшении симптомов и смертности у пациентов с этим заболеванием.[3][7][18] Было показано, что длительное лечение ингибитором ФДЭ5 усиливает путь натрийуретический пептид-цГМФ, снижает уровень Ca2+ сигнальный путь и изменение сосудистого тонуса в легочных артериях в моделях крыс.[9]
Доброкачественная гиперплазия предстательной железы
С 2011 г. препарат длительного действия тадалафил лицензирован для лечения мочевых симптомов, вызванных доброкачественной гиперплазией простаты.[3]
Будущие показания к применению ингибиторов ФДЭ5
Сердечно-сосудистые заболевания
Ингибиторы PDE5 имеют широкий спектр эффектов на сердечно-сосудистую систему, помимо их острого гемодинамического влияния. Например, было показано, что ингибиторы ФДЭ5 улучшают несколько параметров функции эндотелия.[3] Все чаще их использование в управлении системными гипертония (включая резистентную к лечению гипертензию), кардиозащита, сердечная недостаточность, и заболевание периферических артерий оцениваются.[3]
Сердечная недостаточность
Ингибиторы ФДЭ5 показали себя многообещающими при лечении сердечная недостаточность со сниженной фракцией выброса за счет нескольких положительных эффектов на легкие сосудистая сеть, ремоделирование сердца и диастолическая функция.[3] Исследование показало, что эффективное лечение легочной артериальной гипертензии с силденафил улучшение функциональной способности и уменьшение массы правого желудочка у пациентов. Воздействие на ремоделирование правого желудочка было значительно больше по сравнению с неселективным антагонистом эндотелиальных рецепторов. бозентан.[7] Однако ингибиторы ФДЭ5 могут быть вредными для пациентов с сердечной недостаточностью с сохраненной фракцией выброса из-за потенциально отрицательного инотропный последствия.[3]
Хроническая болезнь почек
Экспериментальные исследования на животных показали, что ингибиторы ФДЭ5 могут обратить вспять почка повреждение независимо от их воздействия на артериальное давление через внутрипочечные механизмы.[3] У людей ингибиторы ФДЭ5 также снижают протеинурия, маркер поражения почек.[3] Однако успешное внедрение SGLT2 ингибиторы и антагонисты рецепторов эндотелина в области почечной терапии делает маловероятным разработку ингибиторов ФДЭ5 для этой цели.[3]
Сахарный диабет
Было показано, что ингибиторы PDE5 обладают различными макрососудистый, микрососудистый и метаболический преимущества в сахарный диабет,[3] и в большом исследовании мужчин с сахарный диабет 2 типа Было обнаружено, что агенты значительно снижают риск смерти пациентов от любой причины.[19] Неясно, в какой степени это наблюдение отражает защитные эффекты ингибиторов ФДЭ5 против сердечно-сосудистых и почечных заболеваний.[3]
Феномен Рейно
Силденафил было показано, что он по крайней мере так же эффективен, как блокаторы кальциевых каналов в лечении тяжелых Феномен Рейно (RP), связанный с системным склерозом и язвой пальцев.[3] При приеме силденафила в течение 4 недель у субъектов снизилась средняя частота и продолжительность приступов Рейно и значительно снизился средний балл состояния Рейно. В капилляр Скорость кровотока также увеличивалась у каждого отдельного пациента, а средняя скорость капиллярного потока у всех пациентов значительно увеличивалась. Эти результаты не привели к значительному снижению системного артериальное давление.[7] Однако терапевтические эффекты ингибиторов ФДЭ5 при первичной (идиопатической) РП менее четко определены.[3]
Гладить
Силденафил Было показано, что он значительно улучшает нервно-сосудистую связь, не влияя на общий церебральный кровоток, за счет повышения уровня цГМФ в головном мозге, вызывая нейрогенез и уменьшая неврологический дефицит у крыс через 2 или 24 часа после инсульта. Эти экспериментальные данные предполагают, что ингибиторы PDE5 могут играть роль в ускорении восстановления после Инсульт.[7][9][11] Однако исследования на людях остаются безрезультатными.[3]
Преждевременная эякуляция
Добавление ингибиторов ФДЭ5 к СИОЗС Согласно недавним исследованиям, препараты (например, пароксетин) для лечения преждевременной эякуляции могут улучшить контроль эякуляции.[11] Возможный механизм основан на оксид азота Система трансдукции (NO) / цГМФ как центральный и периферический медиатор тормозящей неадренергической, нехолинергической нитрергической нейротрансмиссии в мочеполовой системе.[16]
Расстройство женского сексуального возбуждения
PDE5 экспрессируется в кавернозном теле клитора, а также в гладких мышцах и эпителии влагалища. Следовательно, возможно, что ингибиторы ФДЭ5 могут влиять на расстройство сексуального возбуждения у женщин, но необходимы дальнейшие исследования. Было показано, что повышенные уровни цГМФ возникают в культивируемых человеком клетках гладких мышц влагалища, обработанных ингибитором ФДЭ5, что предполагает участие оси NO / цГМФ в женской сексуальной реакции.[11]
Расстройство сексуального истощения
Сходство многих ингибиторов ФДЭ5 со структурой многих аналогов кофеин которые также антагонисты аденозина предполагает, что в будущем можно будет разработать ингибитор PDE5, который, как и кофеин, также является антагонистом аденозина.
Открытие
PDE5 - это фермент, который впервые был очищен в 1980 году из легких крыс.[20] PDE5 превращает внутриклеточный цГМФ в нуклеотид GMP.[21] Многие ткани содержат PDE5, такие как легкие, почки, мозг, тромбоциты, печень, простата, уретра, мочевой пузырь и гладкие мышцы. Из-за локализации PDE5 в гладкой мышечной ткани были разработаны ингибиторы для лечения Эректильная дисфункция вместе с легочная гипертония.[1][2]
Силденафил был впервые представлен для клинических испытаний в 1989 году. Это был результат обширных исследований химических агентов, нацеленных на ФДЭ5, которые могут быть эффективны при лечении ишемическая болезнь сердца.[22] Силденафил не оказался эффективным при ишемической болезни сердца, но был обнаружен интересный побочный эффект - половой член. эрекция. Этот побочный эффект вскоре стал основной областью исследования.[23] Ингибитор очень селективен по отношению к семейству PDE5.[22]
Силденафил - это прототип ингибиторов ФДЭ5, которые Pfizer запущен как Виагра. Он был одобрен Управление по контролю за продуктами и лекарствами (FDA) в 1998 году как первая устный лекарство от эректильной дисфункции. Позже, в 2005 году, он был одобрен для лечения легочной артериальной гипертензии.[2] Варденафил и тадалафил были открыты в 1990 году. Эти препараты появились в результате исследовательских программ, направленных на поиск ингибиторов ФДЭ5 для лечения сердечно-сосудистых заболеваний и эректильной дисфункции. Два ингибитора PDE5 вскоре стали лечить эти состояния.[22][23]
Тадалафил является наиболее универсальным ингибитором и имеет самый длительный период полувыведения - 17,5 часов. Это обеспечивает более длительное терапевтическое окно и поэтому часто оказывается более удобным лекарством, чем другие препараты с более коротким терапевтическим интервалом. Тадалафил более биодоступен (80%), чем силденафил (40%) и варденафил (15%), но он имеет медленную абсорбцию, или около 2 часов по сравнению с 50 минутами силденфила. Варденафил наиболее известен своей эффективностью.[24]
Из-за серьезных побочных эффектов и неудовлетворенности пациентов текущим выбором терапии другие ингибиторы недавно были одобрены для клинического использования. Эти ингибиторы - уденфил, аванафил, лоденафил и мироденафил.[25]
Разработка
Биологическая активность
Эрекция полового члена

Эрекция полового члена - это гемодинамический событие в гладкая мышца кавернозного тела.[26] PDE5 является основным ферментом гидролиза цГМФ, обнаруженным в кавернозном теле полового члена.[27] Эрекция вызывается выпуском нейротрансмиттер оксид азота (NO) от неадренергических и нехолинергических нейронов от нервных окончаний в половом члене, а также от эндотелиальные клетки. NO активирует растворимые гуанилилциклаза в гладкомышечных клетках полового члена, что приводит к увеличению производства 3'-5'-циклического гуанозинмонофосфата из гуанозин-5'-трифосфата (GTP).[21][28][29] Циклический GMP связывается с цГМФ-зависимая протеинкиназа (PKG1), который фосфорилирует несколько белков, что приводит к снижению внутриклеточного кальция. Более низкий уровень внутриклеточного кальция приводит к расслаблению гладких мышц и, в конечном итоге, к эрекции полового члена. Этот путь продемонстрирован в Рисунок 1.[29][30]
{kind=link}
Эректильная дисфункция
PDE5 разрушает цГМФ и, следовательно, подавляет эрекцию. Как показано в Рисунок 1, ингибирование PDE5 снижает деградацию цГМФ и приводит к эрекции полового члена.[28][31]Благодаря этому действию ингибиторы PDE5 были разработаны для лечения эректильной дисфункции полового члена.[32]
Фермент фосфодиэстераза 5

Эта подпись использует банальная формулировка. (Июль 2017 г.) |
Фермент PDE5 имеет молекулярная масса 200 кДа и его активное состояние гомодимер.[21] PDE5 состоит из мономеры и каждый содержит два основных функциональных домена: регуляторный домен (R-домен), который расположен в N-концевой части белка, и каталитический домен (C-домен), расположенный в более C-концевой части белка.[33][21]
Домен R содержит специфический аллостерический сайт связывания цГМФ, который контролирует функцию ферментов. Этот специфический сайт связывания состоит из субдомена GAF (цГМФ-специфическая цГМФ-стимулированная ФДЭ, аденилатциклаза и FhlA), который расположен в N-концевом участке специфических белков. Аллостерический сайт связывания GAF состоит из GAFa и GAFb, где GAFa имеет более высокую аффинность связывания. Важность и функциональная роль двух гомологичных сайтов связывания неизвестны.[34]
Конформационное изменение происходит, когда цГМФ связывается с аллостерическим сайтом, который открывает серин и разрешает фосфорилирование. Результаты фосфорилирования серина приводят к усилению гидролиза цГМФ в каталитическом домене. Сродство каталитического домена к цГМФ увеличивается и дополнительно увеличивает активность каталитического домена PDE5.[33]Через домен C внутриклеточный цГМФ быстро разлагается PDE5, что минимизирует активность цГМФ на его субстрате PKG1 за счет отщепления циклической фосфатной части цГМФ до GMP. GMP - неактивная молекула, не имеющая активности вторичного мессенджера.[33][35]
Фосфорилирование одиночного серина PKG1 и аллостерическим сайтом связывания cGMP активирует каталитическую активность PDE5, и в результате негативный отзыв регуляция передачи сигналов cGMP / NO / PKG1. Таким образом, цГМФ взаимодействует как с аллостерическим, так и с каталитическим доменом фермента ФДЭ5, а ингибиторы ФДЭ5 конкурируют с цГМФ за связывание с каталитическим доменом, что приводит к более высоким уровням цГМФ.[33] Домены PDE5 представлены в фигура 2.
{kind=link}
Ингибиторы PDE5

Эта подпись использует банальная формулировка. (Июль 2017 г.) |

Эта подпись использует банальная формулировка. (Июль 2017 г.) |
Ингибиторы ФДЭ5 силденафил, варденафил и тадалафил являются конкурентными и обратимыми ингибиторами гидролиза цГМФ каталитической стороной ФДЭ5. Структуры варденафила и силденафила схожи, они оба содержат схожие по структуре пурин кольцо цГМФ, которое способствует их свойствам действовать как конкурентный ингибитор PDE5. Различие молекулярных структур является причиной взаимодействия с каталитическим сайтом PDE5 и улучшает сродство этих соединений по сравнению с селективностью цГМФ.[33]
Фармакофор
Фармакофорная модель PDE5 обычно состоит из одного акцептора водородной связи, одной гидрофобной алифатической углеродной цепи и двух ароматических колец. Небольшой гидрофобный карман и H-петля фермента PDE5 важны для аффинности связывания ингибиторов PDE5. Во многих случаях при связывании ингибитора наблюдаются позиционные и конформационные изменения.[36]
Активный сайт PDE5 расположен в домене спирального пучка в центре домена C (каталитический домен). Карман субстрата состоит из четырех субсайтов: M-сайт (участок связывания металла), Q-карман (сердцевинный карман), H-карман (гидрофобный карман) и L-область (область крышки), как показано на фигура 3.[37] Карман Q вмещает пиразолопиримидиноновую группу силденафила. Это предполагает, что другие химические вещества, подобные гуанидиновым группам цГМФ, также могут связываться в этой области. Аминокислотные остатки Gln817, Phe820, Val782 и Tyr612 расположены в кармане Q, они высококонсервативны во всех PDE. Амидный фрагмент пиразолопиримидиноновой группы образует бидентатную водородную связь с ɣ-амидной группой Gln817.[37] Трехмерная структура силденафила показана на фигура 4.
{kind=link}
{kind=link}
Побочные эффекты
Ингибиторы ФДЭ5 обычно хорошо переносятся, с побочными эффектами, включая преходящие головные боли, приливы, диспепсию, заложенность и головокружение.[3] Также были сообщения о временных нарушениях зрения при приеме силденафила и, в меньшей степени, варденафила, а также о болях в спине и мышцах при приеме тадалафила.[3] Эти побочные эффекты могут быть отнесены к непредвиденным эффектам ингибиторов PDE5 против других изоферментов PDE, таких как PDE1, PDE6 и PDE11. Предполагается, что улучшенная селективность ингибиторов PDE5 может привести к меньшему количеству побочных эффектов.[3] Например, варденафил и тадалафил продемонстрировали снижение побочных эффектов, вероятно, из-за улучшенной селективности в отношении PDE5.[38] Однако в настоящее время высокоселективные ингибиторы PDE5 не разрабатываются.[3]
Пациенты, принимающие нитраты, альфа-блокаторы или стимуляторы рГЦ в течение 24 часов после введения ингибитора ФДЭ5 (или 48 часов для тадалафила) могут иметь симптомы гипотония, поэтому одновременное применение противопоказано.[3] Ингибиторы PDE5 также противопоказаны пациентам с наследственными заболеваниями глаз, такими как пигментный ретинит из-за небольшого повышенного риска неартеритического ишемическая оптическая нейропатия у пациентов, принимающих лекарство.[3]
Нарушение слуха является одним из факторов риска для тех, кто принимает ингибиторы ФДЭ5, и было зарегистрировано для всех доступных на рынке лекарств. Эта проблема может быть связана с высоким уровнем воздействия цГМФ на волосковые клетки улитки.[33] Сообщалось, что ингибиторы ФДЭ5 (силденафил и варденафил) вызывают временные нарушения зрения, вероятно, из-за ингибирования ФДЭ6.[3]
Несколько отчетов касаются подходов к улучшению ингибиторов PDE5, когда химические группы были заменены для повышения эффективности и селективности, что потенциально должно привести к лекарствам с меньшим количеством побочных эффектов.[38][39]
Взаимосвязь между структурой и деятельностью (SAR)
Силденафил, первый ингибитор ФДЭ5, был открыт в рамках программы рациональной разработки лекарств. Соединение было сильнодействующим и селективным по сравнению с PDE5, но не имело предпочтительных фармакологических свойств.[40]
Связь структура-деятельность (SAR) демонстрируется в цифра 5, рисунок 6 и рисунок 7. Рисунок 5 демонстрирует три основные группы силденафила, R1, R2 и R3. R1 представляет собой пиразолопиримидиноновое кольцо, R2 - этоксифенильное кольцо, а R3 - метилпиперазиновое кольцо. Группа R1 отвечает за связывание лекарства с его активным сайтом связывания PDE5.[27]

Эта подпись использует банальная формулировка. (Июль 2017 г.) |
Растворимость - одно из повышенных фармакологических свойств. Группа была заменена на атом водорода, как показано на рисунок 6. В сульфонамид группа была выбрана для снижения липофильности и увеличения растворимости, как показано на рисунок 7.[1][39]

Эта подпись использует банальная формулировка. (Июль 2017 г.) |
Растворимость была дополнительно увеличена путем помещения метильная группа в позициях R, как показано на рисунок 7. Другие ингибиторы фосфодиэстеразы-5 были разработаны на основе структуры в рисунок 7.[1][39]

Эта подпись использует банальная формулировка. (Июль 2017 г.) |
Другое исследование
Хотя основное применение ингибиторов ФДЭ5 было при эректильной дисфункции, большой интерес к ингибиторам ФДЭ5 как к многообещающим новым терапевтическим агентам для лечения других заболеваний, таких как Болезнь Альцгеймера. Повышение уровня цГМФ за счет ингибирования ФДЭ5 обеспечивает способ улучшения памяти и обучения.[1]PDE5 также рассматривается как потенциальный терапевтический агент для паразитарная болезнь Такие как Африканская сонная болезнь. В структуру силденафила были внесены стратегические изменения, чтобы молекула могла проецироваться в специфический карман для паразитов (p-карман). Аналогичный подход был использован для разработки терапевтических агентов. Плазмодий falciparum.[2]
Ингибиторы ФДЭ5 в клинических испытаниях
| Препарат, средство, медикамент | Статус клинического исследования (2005 г.) | Индикация | Режиссер |
|---|---|---|---|
| UK357903 | Фаза II | Эректильная дисфункция (ингибитор ФДЭ5 второго поколения)[9] | Pfizer |
| Аванафил | Фаза II | Эректильная дисфункция и расстройство женского сексуального возбуждения[9] | Танабэ |
| Уденафил (DA-8159) | Фаза II | Эндотелиальная дисфункция,[9] Эректильная дисфункция[9] и эректильная дисфункция, связанная с ожирение,[41] сахарный диабет[42] и использование СИОЗС[43] | Dong-A Pharmaceutical |
Смотрите также
Рекомендации
- ^ а б c d е ж Fiorito, J .; Zhang, H .; Станишевский, А .; Feng, Y .; Фрэнсис, Ю. И. (2013). «Синтез производных хинолина: открытие мощного и селективного ингибитора фосфодиэстеразы 5 для лечения болезни Альцгеймера». Eur J Med Chem. 60: 285–294. Дои:10.1016 / j.ejmech.2012.12.009. ЧВК 3582828. PMID 23313637.
- ^ а б c d Wang, G .; Liu, Z .; Chen, T .; Wang, Z .; Ян, H .; Чжэн, М .; Цзян, Х. (2012). «Дизайн, синтез и фармакологическая оценка моноциклических пиримидинонов как новых ингибиторов PDE5». J Med Chem. 55 (23): 10540–10550. Дои:10.1021 / jm301159y. PMID 23137303.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс Тзумас, Николаос; Фарра, Тарик Э .; Дхаун, Нирадж; Уэбб, Дэвид Дж. (12 ноября 2019 г.). «Установленные и новые терапевтические применения ингибиторов фосфодиэстеразы 5 типа при сердечно-сосудистых заболеваниях». Британский журнал фармакологии. Дои:10.1111 / бф.14920. ISSN 1476-5381. PMID 31721165.
- ^ а б Bingham, J .; Сударшанам С. и Шринивасан С. (2006). «Профилирование генов фосфодиэстеразы человека и изоформ сплайсинга».Сообщения о биохимических и биофизических исследованиях 350, 25-32.
- ^ а б Jiang, W. Q .; и другие. (2004). «Профилирование синтеза и SAR тетрациклических пирролохинолонов как ингибиторов фосфодиэстеразы 5». Биоорганическая и медицинская химия 12, 1505-1515.
- ^ Гарретт (2002). Принципы биохимии: с фокусом на человека. Форт-Уэрт: издатели колледжа Харкорт. ISBN 978-0-03-097369-7.
- ^ а б c d е ж грамм Ghofrani, H.A .; Остерло, И. Х. и Гриммингер, Ф. (2006). «Силденафил: от стенокардии и эректильной дисфункции до легочной гипертензии и не только». Nature Reviews Drug Discovery 5, 689-702.
- ^ а б Sung, B.J .; и другие. (2003). «Структура каталитического домена человеческой фосфодиэстеразы 5 со связанными молекулами лекарственного средства». Nature 425, 98-102.
- ^ а б c d е ж грамм час я j k л Jeon, Y.H .; и другие. (2005). «Фосфодиэстераза: обзор белковых структур, потенциальных терапевтических применений и последних достижений в разработке лекарств». Cmls-Cellular and Molecular Life Sciences 62, 1198-1220.
- ^ а б Лин, С. С. (2004). «Тканевая экспрессия, распределение и регуляция PDE5». Международный журнал исследований импотенции 16, S8-S10.
- ^ а б c d е Джексон, G .; Гиллис, Х. и Остерло, И. (2005). «Прошлое, настоящее и будущее: 7-летнее обновление Виагры ((R)) (цитрат силденафила)». Международный журнал клинической практики 59, 680-691.
- ^ Blount, M. A .; и другие. (2004). «Связывание меченного тритием силденафила, тадалафила или варденафила с каталитическим сайтом фосфодиэстеразы-5 демонстрирует эффективность, специфичность, гетерогенность и стимуляцию цГМФ». Молекулярная фармакология 66, 144-152.
- ^ Лунье, К. (2006). «Суперсемейство циклических нуклеотидфосфодиэстераз (PDE): новая мишень для разработки конкретных терапевтических агентов». Фармакология и терапия 109, 366-398.
- ^ а б c d е ж грамм Карточка, Г. Л .; и другие. (2004). «Структурные основы действия препаратов, подавляющих фосфодиэстеразы». Структура 12, 2233-2247.
- ^ Chen, J .; и другие. (2003). "MMDB: база данных 3D-структуры Entrez 10.1093 / nar / gkg086". Nucleic Acids Res. 31, 474-477.
- ^ а б McMahon, C.G .; McMahon, C.N .; Леоу, Л. Дж. И Винсток, К. Г. (2006). «Эффективность ингибиторов фосфодиэстеразы 5 типа в медикаментозном лечении преждевременной эякуляции: систематический обзор». Bju International 98, 259-272.
- ^ Shinlapawittayatorn, K .; Чаттипакорн, С. & Чаттипакорн, Н. (2005). «Влияние цитрата силденафила на сердечно-сосудистую систему». Бразильский журнал медико-биологических исследований 38, 1303-1311.
- ^ Чунг, К. Ф. (2006). «Ингибиторы фосфодиэстеразы при заболеваниях дыхательных путей». Европейский журнал фармакологии 533, 110-117.
- ^ Андерсон, Саймон Дж .; Хатчингс, Дэвид С.; Вудворд, Марк; Рахими, Казем; Раттер, Мартин К .; Кирби, Майк; Хакетт, Джефф; Траффорд, Эндрю В .; Хилд, Адриан Х. (01.11.2016). «Использование ингибиторов фосфодиэстеразы 5 типа при диабете 2 типа связано со снижением общей смертности». Сердце. 102 (21): 1750–1756. Дои:10.1136 / heartjnl-2015-309223. ISSN 1355-6037. ЧВК 5099221. PMID 27465053.
- ^ Francis, S. H .; Lincoln, T. M .; Корбин, Дж. Д. (1980). «Характеристика нового связывающего цГМФ белка из легких крысы». Журнал биологической химии. 255 (2): 620–626. PMID 6153179.
- ^ а б c d Ротелла, Д. П. (2002). «Ингибиторы фосфодиэстеразы 5: текущее состояние и возможности применения». Обзоры природы. Открытие наркотиков. 1 (9): 674–682. Дои:10.1038 / nrd893. PMID 12209148.
- ^ а б c Ravipati, G .; McClung, J. A .; Aronow, W. S .; Петерсон, С. Дж .; Фришман, В. Х. (2007). «Ингибиторы фосфодиэстеразы 5 типа в лечении эректильной дисфункции и сердечно-сосудистых заболеваний». Кардиол Рев. 15 (2): 76–86. Дои:10.1097 / 01.crd.0000233904.77128.49. PMID 17303994.
- ^ а б Reffelmann, T .; Клонер, Р. А. (2003). «Терапевтический потенциал ингибирования фосфодиэстеразы 5 при сердечно-сосудистых заболеваниях». Тираж. 108 (2): 239–244. Дои:10.1161 / 01.CIR.0000081166.87607.E2. PMID 12860892.
- ^ Киркпатрик, П; Ноймайер, К. (2004). «Тадалафил и варденафил». Natural Ref Drug Discovery. 3 (4): 295–296. Дои:10.1038 / nrd1362. PMID 15124623.
- ^ Kedia, G.T .; Uckert, S .; Assadi-Pour, F .; Кучик, М.А. (2013). «Аванафил для лечения эректильной дисфункции: исходные данные и основные клинические свойства». Ther Adv Urol. 5 (1): 35–41. Дои:10.1177/1756287212466282. ЧВК 3547533. PMID 23372609.
- ^ Chen, C. Y .; Chang, Y.H .; Bau, D. T .; Huang, H.J .; Tsai, F.J .; Tsai, C.H .; Чен, К. Ю. (2009). «Открытие мощных ингибиторов фосфодиэстеразы 5 путем виртуального скрининга и фармакофорного анализа». Акта Фармакол Син. 30 (8): 1186–1194. Дои:10.1038 / aps.2009.100. ЧВК 4006686. PMID 19597523.
- ^ а б Пиво, Д .; Bhalay, G .; Dunstan, A .; Glen, A .; Haberthuer, S .; Moser, H .; Цзян, Х. (2002). «Твердофазный подход к синтезу ингибиторов PDE5». Биоорг Мед Хем Летт. 12 (15): 1973–1976. Дои:10.1016 / S0960-894X (02) 00296-2. PMID 12113821.
- ^ а б Бернетт, А. Л. (2006). «Роль оксида азота в эректильной дисфункции: значение для медицинской терапии». J Clin Hypertens (Гринвич). 8 (12): 53–62. Дои:10.1111 / j.1524-6175.2006.06026.x. PMID 17170606.
- ^ а б Корбин, Дж. Д. (2004). «Механизмы действия ингибирования ФДЭ5 при эректильной дисфункции». Int J Impot Res. 16 (1): 4–7. Дои:10.1038 / sj.ijir.3901205. PMID 15224127.
- ^ Андерссон, К. (2001). «Фармакология эрекции полового члена». Фармакологические обзоры. 53 (3): 417–50. PMID 11546836.
- ^ Коул, Хари; Bivalacqua, Trinity J .; Musicki, Biljana; Hsu, Lewis L .; Берковиц, Дэн Э .; Чемпион, Охотник К .; Бернетт, Артур Л. (2013). "Силденафил-цитрат-восстановленная регуляция eNOS и PDE5 в пенисе серповидно-клеточной мыши предотвращает приапизм посредством контроля окислительного / нитрозативного стресса". PLOS ONE. 8 (7): e68028. Bibcode:2013PLoSO ... 868028B. Дои:10.1371 / journal.pone.0068028. ISSN 1932-6203. ЧВК 3699477. PMID 23844149.
- ^ Shamloul, R .; Ганем, Х. (2013). "Эректильная дисфункция". Ланцет. 381 (9861): 153–165. Дои:10.1016 / S0140-6736 (12) 60520-0. PMID 23040455.
- ^ а б c d е ж Cockrill, B.A .; Ваксман, А. Б. (2013). Ингибиторы фосфодиэстеразы-5. J Med Chem. Справочник по экспериментальной фармакологии. 218. С. 229–255. Дои:10.1007/978-3-642-38664-0_10. ISBN 978-3-642-38663-3. PMID 24092343.
- ^ Турко, И. В .; Francis, S. H .; Корбин, Дж. Д. (1998). «Связывание цГМФ с обоими аллостерическими сайтами цГМФ-связывающей цГМФ-специфической фосфодиэстеразы (ФДЭ5) необходимо для его фосфорилирования». Биохимический журнал. 329 (3): 505–510. Дои:10.1042 / bj3290505. ЧВК 1219070. PMID 9445376.
- ^ Окада, Д .; Асакава, С. (2002). «Аллостерическая активация cGMP-специфической, cGMP-связывающей фосфодиэстеразы (PDE5) с помощью cGMP». Биохимия. 41 (30): 9672–9679. Дои:10.1021 / bi025727 +. PMID 12135389.
- ^ Tomori, T .; Hajdu, I .; Lorincz, Z .; Cseh, S .; Дорман, Г. (2012). «Комбинирование методов 2D и 3D in silico для быстрого выбора потенциальных ингибиторов PDE5 из хранилищ многомиллионных соединений: биологическая оценка». Мол Дайверс. 16 (1): 59–72. Дои:10.1007 / s11030-011-9335-0. PMID 21947759.
- ^ а б Sung, B.J .; Hwang, K. Y .; Jeon, Y.H .; Lee, J. I .; Heo, Y. S .; Ким, Дж. Х. (2003). «Структура каталитического домена человеческой фосфодиэстеразы 5 со связанными молекулами лекарственного средства». Природа. 425 (6953): 98–102. Bibcode:2003Натура 425 ... 98С. Дои:10.1038 / природа01914. PMID 12955149.
- ^ а б Ю., Г. Х .; Мейсон, H .; Wu, X. M .; Wang, J .; Chong, S. H .; Бейер, Б. (2003). «Замещенные пиразолопиридопиридазины как пероральные биодоступные сильные и селективные ингибиторы PDE5: потенциальные средства для лечения эректильной дисфункции». J Med Chem. 46 (4): 457–460. Дои:10.1021 / Jm0256068. PMID 12570368.
- ^ а б c Писсарницкий, Д. А .; Asberom, T .; Boyle, C.D .; Chackalamannil, S .; Чинтала, М .; Clader, J. W .; Сюй Р. (2004). «Разработка SAR полициклических производных гуанина, направленная на открытие селективного ингибитора PDE5 для лечения эректильной дисфункции». Биоорг Мед Хем Летт. 14 (5): 1291–1294. Дои:10.1016 / j.bmcl.2003.12.027. PMID 14980684.
- ^ Кэмпбелл, С.Ф. (2000). «Наука, искусство и открытие лекарств: личная перспектива». Клиническая наука. 99 (4): 255–260. Дои:10.1042 / cs20000140. PMID 10995589.
- ^ Yu, J. Y .; Канг, К. К. и Ю, М. (2006). «Эректильные возможности нового ингибитора фосфодиэстеразы 5 типа, DA-8159, у крыс с ожирением, вызванным диетой». Азиатский журнал андрологии 8, 325-329.
- ^ Ahn, G.J .; и другие. (2005). «Хроническое введение ингибитора фосфодиэстеразы 5 улучшает эректильную и эндотелиальную функцию на крысиной модели диабета». Международный журнал андрологии 28, 260-266.
- ^ Ahn, G.J .; и другие. (2005). «DA-8159 обращает вспять эректильную дисфункцию, вызванную селективным ингибитором обратного захвата серотонина у крыс». Урология 65, 202-207.