Открытие и разработка прямых ингибиторов Ха - Discovery and development of direct Xa inhibitors
Четыре препарата из класса прямых ингибиторов Ха продаются во всем мире. Ривароксабан (Xarelto) был первым одобренным ингибитором FXa, который стал коммерчески доступным в Европе и Канаде в 2008 году.[1] Второй был апиксабан (Eliquis), утвержден в Европе в 2011 г.[2] и в США в 2012 году.[3] Третий Эдоксабан (Lixiana, Savaysa) была одобрена в Японии в 2011 году и в Европе и США в 2015 году.[4] Бетриксабан (Bevyxxa) был одобрен в США в 2017 году.
История
Гепарин
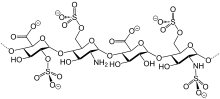
Гепарин был обнаружен Джей Маклин и Уильям Генри Хауэлл в 1916 году он был впервые выделен из печени собаки, что в переводе с греческого означает печень. Гепарин нацелен на несколько факторов в каскаде свертывания крови, одним из которых является FXa. Сначала у него было много побочных эффектов, но в течение следующих двадцати лет исследователи работали над гепарином, чтобы сделать его лучше и безопаснее. Он вошел клинические испытания в 1935 году, а первое лекарство было выпущено в 1936 году. Цепи природного гепарина могут варьироваться от 5.000 до 40.000 Дальтон. В 80-е годы Низкомолекулярный гепарин (НМГ) были разработаны, и они содержат только цепи со средней молекулярной массой менее 8.000 Да.[5]
Варфарин

В 1920-х годах в Канаде и на севере США произошла вспышка загадочной геморрагической болезни крупного рогатого скота. Заболевание было названо болезнью донника, потому что крупный рогатый скот пасся на сене донника. Лишь через десять лет после вспышки местный следователь, Карл П. Линк и его ученик Вильгельм Шеффель начали интенсивное расследование, чтобы найти вещество, вызывающее внутреннее кровотечение. Им потребовалось 6 лет, чтобы обнаружить дикумарол, вызывающий агент.[5] Они запатентовали право на это вещество, и в 1945 году Линк начал продавать производное кумарина в качестве родентицид. Он и его коллеги работали над несколькими вариантами и в итоге получили вещество, которое они назвали варфарин в 1948 году. Только в 1954 году он был одобрен для использования в медицине у людей, что сделало варфарин первым пероральным антикоагулянтный препарат.[6]
Потребность в новых и лучших пероральных препаратах
Лечение варфарином требует регулярного мониторинга крови и корректировки дозы из-за его узкого терапевтическое окно. При недостаточном контроле варфарин может вызвать слишком частые геморрагические события и многократное взаимодействие с пищей и другими лекарствами. В настоящее время основной проблемой низкомолекулярного гепарина (НМГ) является способ введения, поскольку он должен вводиться подкожно.[7] Из-за этих недостатков возникла острая необходимость в улучшенных антикоагулянтных препаратах. Для современного общества удобный и быстрый прием лекарств - залог хорошего комплаенс. В 2008 году первый прямой ингибитор Ха был одобрен для клинического применения.[8] Прямые ингибиторы Ха столь же эффективны, как НМГ и варфарин, но они назначаются перорально и не нуждаются в таком строгом контроле.[7] Другими преимуществами ингибиторов Ха являются быстрое начало действия / исчезновение, небольшое количество лекарственных взаимодействий и предсказуемость. фармакокинетика. Быстрое начало / компенсация эффекта значительно снижает потребность в «переходе» с парентеральными антикоагулянтами после операций.[9] Сегодня на рынке представлены четыре ингибитора фактора Ха: ривароксабан, апиксабан, Эдоксабан и Betrixaban.[7]
Антистазин и клещевой антикоагулянтный пептид (ТАП)
Фактор Ха был определен как многообещающая цель для разработки новых антикоагулянтов в начале 1980-х годов. В 1987 году первый ингибитор фактора Ха, природный комплексный антистазин, был выделен из слюнные железы мексиканской пиявка Haementeria officinalis. Антистазин - это полипептид и мощный ингибитор Ха. В 1990 году был выделен еще один природный ингибитор Ха - антикоагулянтный пептид клещей (ТАР) из экстрактов клещей. Ornithodoros moubata. ТАР и антистазин использовались для оценки фактора Ха как мишени для лекарства.[8]
Механизм действия

Коагуляция крови это сложный процесс, при котором кровь образует сгустки. Это важная часть гемостаз и работает, останавливая потерю крови из поврежденных кровеносных сосудов.[10] В месте травмы, где есть обнажение крови под эндотелий, тромбоциты собираются и сразу же образуют пробку. Этот процесс называется первичным гемостазом. Одновременно происходит вторичный гемостаз. Он определяется как образование нерастворимых фибрин активированными факторами свертывания, в частности тромбином.[11] Эти факторы активируют друг друга в каскаде свертывания крови, который происходит посредством двух отдельных взаимодействующих путей: внутреннего и внешнего.[12] После активации различных проферментов на последних этапах каскада образуется тромбин, который затем преобразует фибриноген в фибрин, который приводит к образованию сгустков.[10] Фактор Ха представляет собой активированную сериновую протеазу, которая играет ключевую роль в пути свертывания крови, превращая протромбин в тромбин. Ингибирование фактора Ха приводит к антитромботическим эффектам за счет уменьшения количества тромбина. Предполагается, что прямое воздействие на фактор Ха является эффективным подходом к антикоагуляции.[8]
Разработка

В 1987 году антистазин был испытан как первый прямой ингибитор Ха. Антистазин - это белок, состоящий из 119 аминокислотных остатков, из которых 20 являются цистеины участвует в 10 дисульфидные связи.[13] Он действует как медленный, прочный ингибитор фактора Ха с Значение Ki 0,3–0,6 нМ, но также ингибирует трипсин.[8] Рекомбинантный антистазин можно производить с помощью генетически модифицированных дрожжей, saccharomyces cerevisiae.[14] Другой природный прямой ингибитор Ха, клещевой антикоагулянтный пептид (ТАП), был открыт в 1990 году. Это одноцепочечный пептид из 60 аминокислот и, как и антистазин, медленный, прочно связывающийся ингибитор с аналогичным значением Ki ( ~ 0,6 нМ).
Эти два белка в основном использовались для проверки фактора Ха как фактора мишень для наркотиков. Исследования на животных показали, что прямое ингибирование Ха является более эффективным подходом к антикоагуляции по сравнению с прямыми ингибиторами тромбина, особенно предлагая более широкий терапевтическое окно и снижение риска отскока тромбоз, (увеличить в тромбоэмболические события возникающие вскоре после отмены антитромботических препаратов) по сравнению с прямым и непрямым ингибиторы тромбина.[8]
В течение 1990-х годов было разработано несколько низкомолекулярных веществ, таких как DX-9065a.[15] и YM-60828.[16]

DX-9065a был первым синтетическим соединением, которое ингибировало FXa без ингибирования тромбина. Это было достигнуто путем вставки карбоксильная группа который, по-видимому, является наиболее важным фрагментом для избирательного связывания с FXa.[8] Те ранние развитые маленькие молекулы еще имел амидин -группы или даже более высокие базовые функции, которые считались необходимыми в качестве имитации для аргинин остаток в протромбин, естественный субстрат фактора Ха. Тем не менее, эти основные функции также связаны с очень плохим оральным биодоступность (например, 2–3% для DX-9065a).
В 1998 г. Bayer Healthcare, фармацевтическая компания начала поиск низкомолекулярных прямых ингибиторов фактора Ха с более высокой пероральной биодоступностью. Скрининг с высокой пропускной способностью и дальнейшая оптимизация сначала привела к нескольким веществам из класса изоиндолинонов, демонстрирующим, что гораздо меньше основных веществ могут также действовать как сильные ингибиторы Ха в отношении IC50 значение до 2 нМ. Хотя изоиндолиноны имеют лучшую пероральную биодоступность, чем исходные соединения, этого было недостаточно. Однако позже проект привел к классу н-арилоксазолидиноны который обеспечивает вещества как с высокой эффективностью ингибирования фактора Ха, так и с высокой биодоступностью.[8] Одно соединение этого класса, Ривароксабан (IC50 = 0,7 нМ, биодоступность: 60%), было выдано разрешение на продажу для предотвращения Венозная тромбоэмболия в Европе и Канаде в сентябре 2008 г.[1][17]
Химия
Фактор Ха: структура и сайты связывания

Факторы IIa, Xa, VIIa, IXa и XIa представляют собой протеолитические ферменты, которые играют определенную роль в каскаде свертывания крови. Фактор Ха (FXa) является наиболее многообещающим из-за его положения на пересечении внутреннего и внешнего пути, а также генерирования около 1000 молекул тромбина для каждой молекулы Ха, что приводит к сильному антикоагулянтному эффекту. FXa образуется из FX путем расщепления активационного пептида из 52 аминокислот, поскольку «a» в факторе Xa означает активированный. FXa состоит из каталитического домена из 254 аминокислот и также связан с легкой цепью из 142 аминокислот. Цепочка содержит как GLA домен и два фактор роста эпидермиса домены (EGF-подобные домены).[18]
Активный сайт FXa структурирован так, чтобы катализировать расщепление физиологических субстратов и расщеплять PhePheAsnProArg-ThrPhe и TyrIleAspGlyArg-IleVal в протромбине. FXa имеет четыре так называемых кармана, которые являются мишенями для связывания субстратов с фактором Xa. Эти карманы выстроены в линию из разных аминокислот, и ингибиторы Ха нацелены на эти карманы при связывании с фактором Ха. Двумя наиболее важными карманами в отношении аффинности и селективности в отношении ингибиторов Ха являются S1 и S4.[18]
S1: Карман S1 представляет собой гидрофобный карман и содержит остаток аспарагиновой кислоты (Asp-189), который может служить сайтом узнавания для основной группы. FXa имеет остаточное пространство в кармане S1 и выстлан остатками Тюр -228, Жерех -189 и Сер-195.[18]
S2: Карман S2 - это небольшой и неглубокий карман. Он сливается с карманом S4 и имеет место для небольших аминокислот. Похоже, Tyr-99 блокирует доступ к этому карману, поэтому этот карман не так важен, как S1 и S4.[19]
S3: Карман S3 расположен на краю кармана S1, плоский и подвержен воздействию растворителя. Этот карман не так важен, как S1 и S4.
S4: Карман S4 является гидрофобным по своей природе, а дно кармана образовано остатком Trp-215. Остатки Phe-174 и Tyr-99 FXa присоединяются к Trp-215 с образованием ароматического бокса, который способен связывать алифатические, ароматические и положительно заряженные фрагменты. Из-за связывания с положительно заряженными объектами его можно описать как катионную дыру.[18]
Химическая структура и свойства прямых ингибиторов Ха
| Ривароксабан | Апиксабан | Эдоксабан | |
|---|---|---|---|
| МВт (г / моль) | 436 | 460 | 548 |
| Молекулярная формула | C19ЧАС18ClN3О5S | C25ЧАС25N5О4 | C24ЧАС30ClN7О4S |
| Форма | L | L | L |
| Kя | 0,4 нМ | 0,08 нМ | 0,561 нМ |
| IC50 | 0,7 нМ | Нет данных | Нет данных |
| Биодоступность при пероральном введении (%) | 66–100 (в зависимости от дозы) | 50 | 62 |

Связывание ингибиторов Ха с фактором Ха
Все ингибиторы Ха связываются так называемым L-образным образом в активном центре фактора Ха. Ключевыми составляющими фактора Ха являются сайты связывания S1 и S4. Сначала было отмечено, что природные соединения, антистазин и ТАП, которые обладают высокополярными и, следовательно, заряженными компонентами, связываются с мишенью с некоторой специфичностью. Вот почему новые лекарства были разработаны с положительно заряженными группами, но они привели к плохой биодоступности. Таким образом, имеющиеся в настоящее время на рынке ингибиторы Ха содержат ароматическое кольцо с различными фрагментами, присоединенными для различных взаимодействий с сайтами связывания S1 и S4. Это также обеспечивает хорошую биодоступность, а также сохранение прочной силы связывания. Ингибиторы Ха, представленные в настоящее время на рынке, поэтому полагаются на гидрофобные и водородные связи, а не на высокополярные взаимодействия.[20]
Связывание антистазина с фактором Ха
Антистазин содержит N- и C-концевые домены, которые похожи по своим аминокислотным последовательностям с ~ 40% идентичностью и ~ 56%. гомология. Каждый из них содержит короткую β-листовую структуру и 5 дисульфидные связи. Только N-концевой домен необходим для ингибирования Ха, в то время как C-терминал домен не вносит вклад в ингибирующие свойства из-за различий в трехмерной структуре, даже несмотря на то, что C-концевой домен имеет сильно аналогичный образец фактическому активному сайту.[13]
Во взаимодействии антистазина с FXa задействованы как активный сайт, так и неактивная поверхность FXa. Реактивный сайт антистазина, образованный Arg-34 и Val-35 в N-концевом домене, соответствует сайту связывания FXa, скорее всего, карману S1. В то же время Glu-15, расположенный за пределами реактивного сайта антистазина, соответствует положительно заряженным остаткам на поверхности FXa. Множественное связывание является термодинамически выгодным и приводит к субнаномолярному ингибированию (Ki = 0,3–0,6 нМ[8]).[13]
Связывание DX-9065a с фактором Ха
DX-9065a, первый низкомолекулярный прямой ингибитор Ха, представляет собой производное амидиноарила с молекулярной массой 571,07 г / моль.[21] Его положительно заряженная амидинонафталиновая группа образует солевой мостик к Жерех -189 остатков в кармане S1 FXa. Кольцо пирролидина помещается между Tyr-99, Phe-174 и Trp-215 в кармане S4 FXa.[22]
В отличие от старых препаратов, например гепарин, DX-9065a, селективен в отношении FXa по сравнению с тромбином, хотя FXa и тромбин похожи по своей структуре. Это вызвано различием в аминокислотном остатке в положении 192 гомолога. В то время как FXa имеет остаток глутамина в этом положении, тромбин имеет глутаминовую кислоту, которая вызывает электростатическое отталкивание с карбоксильной группой DX-9065a. Кроме того, солевой мостик между Glu-97 тромбина и амидиновой группой, закрепленной в пирролидиновом кольце DX-9065a, снижает гибкость молекулы DX-9065a, которая теперь не может достаточно вращаться, чтобы избежать электростатического столкновения. Вот почему IC50 ценить для тромбина> 1000 мкМ, в то время как IC50 значение для FXa составляет 0,16 мкМ.[22]

Связывание ривароксабана с фактором Ха
Связывание ривароксабана с FXa опосредуется двумя водородными связями с аминокислотой Gly-219. Эти две водородные связи играют важную роль, направляя лекарство в субсайты S1 и S4 FXa. Первая водородная связь - это сильное взаимодействие, которое исходит от карбонильного кислорода оксазолидинонового ядра ривароксабана. Вторая водородная связь является более слабым взаимодействием и происходит от аминогруппы хлортиофенкарбоксамидного фрагмента.
Эти две водородные связи приводят к тому, что лекарство образует L-образную форму и помещается в карманы S1 и S4. Аминокислотные остатки Phe-174, Tyr-99 и Trp-215 образуют узкий гидрофобный канал, который является карманом связывания S4. Морфолиноновая часть ривароксабана «зажата» между аминокислотами Tyr-99 и Phe-174, а арильное кольцо ривароксабана ориентировано перпендикулярно по Трп-215. Карбонильная группа морфолинона не имеет прямого взаимодействия с основной цепью FXa, вместо этого она способствует планаризации морфолинонового кольца и, следовательно, способствует размещению ривароксабана между двумя аминокислотами.
Взаимодействие между хлорным заместителем тиофенового фрагмента и ароматическим кольцом Tyr-228, которое расположено в нижней части S1, очень важно из-за того, что оно устраняет необходимость в сильно основных группах для высокого сродства к FXa. Это позволяет ривароксабану, который не является основным, достичь хорошей биодоступности и эффективности при приеме внутрь.[8]

Связывание апиксабана с фактором Ха
Апиксабан демонстрирует такой же способ связывания, что и ривароксабан, и образует прочный комплекс ингибитор-фермент при соединении с FXa. Пара-метоксигруппа апиксабана соединяется с карманом S1 FXa, но, по-видимому, не взаимодействует с какими-либо остатками в этой области FXa. В пиразол N-2 азот атом апиксабана взаимодействует с Gln-192 и карбонил кислород взаимодействует с Gly-216. Фениллактамная группа апиксабана расположена между Tyr-99 и Phe-174 и благодаря своей ориентации способна взаимодействовать с Trp-215 кармана S4. Карбонильная кислородная группа лактамного фрагмента взаимодействует с молекулой воды и, по-видимому, не взаимодействует с какими-либо остатками в кармане S4.[23]

Структура-активность-отношения (SAR)
Важной частью разработки соединения, которое является идеальным ингибитором определенной мишени, является понимание аминокислотной последовательности сайта-мишени, с которой соединение должно связываться. Моделирование протромбина и FXa позволяет определить разницу и идентифицировать аминокислоты в каждом сайте связывания. Внизу кармана S1 на FXa связывающая аминокислота представляет собой Asp-189, с которым могут связываться амидиновые фрагменты. После рентгеновского исследования сайта связывания FXa было обнаружено, что карман S1 имеет плоскую форму, что означает, что плоская амидиноарильная группа должна связываться с ним без стерических препятствий.[8]
Современные прямые ингибиторы Xa представляют собой L-образные молекулы, концы которых идеально помещаются в карманы S1 и S4. Длинная сторона L-образной формы должна соответствовать узкоспециализированному туннелю в активном участке мишени. Для этого эта часть молекул разработана таким образом, чтобы формально не взаимодействовать с FXa в этой области. Поскольку специфического связывания нет, размещение этих агентов между карманами FXa увеличивает общую специфичность лекарств к молекуле FXa. Взаимодействие между карманом S1 FXa и ингибитором может быть как ионным, так и неионным, что важно, поскольку оно позволяет регулировать конструкцию фрагмента для увеличения пероральной биодоступности. Ранее разработанные соединения представляли собой заряженные молекулы, которые плохо всасываются в желудочно-кишечном тракте и поэтому не достигают высоких концентраций в сыворотке. Новые препараты имеют лучшую биодоступность, поскольку они не заряжены и неионно взаимодействуют с карманом S1.[20]
Ривароксабан
Во время разработки ривароксабана с помощью SAR исследователи поняли, что добавление группы 5-хлоротиофен-2-карбоксамида к ядру оксазолидонина может увеличить эффективность в 200 раз, что ранее было слишком слабым для медицинского использования. В дополнение к этому открытию было подтверждено явное предпочтение (S) -конфигурации. Это соединение имело многообещающий фармакокинетический профиль и не содержало высокоосновную амидиновую группу, но ранее считалось важным для взаимодействия с карманом S1. Эти результаты приводят к обширному SAR (взаимосвязь структура-деятельность ) исследования. Во время тестирования SAR R1 был определен как наиболее важная группа для эффективности. Пирролидинон был первой функциональной группой R1, которая значительно увеличила эффективность, но дальнейшие исследования показали даже более высокую эффективность с группой морфолинона. Группы R2 и R3 имели присоединенный водород или фтор, и было быстро установлено, что наличие водорода приводит к наивысшей эффективности. Затем группы R2 и R3 были заменены различными группами, которые были менее активными, чем водород, поэтому водород был конечным результатом. Поскольку хлоротиофеновый фрагмент имел недостаточную растворимость в воде, предпринимались попытки замещения его другой группой, но безуспешно. Фрагмент хлоротиофена связывается с Tyr-228 на дне кармана S1, что делает его ключевым фактором связывания с FXa. Ривароксабан обладает как высоким сродством, так и хорошей биодоступностью.[24]

Апиксабан

Во время разработки апиксабана SAR необходимо было провести тестирование трех групп для достижения максимальной эффективности и биодоступности. Первой тестируемой группой был неактивный сайт, так как он должен быть стабилизирован перед тестированием SAR на п-метоксифенильной группе (S1-связывающий фрагмент). Есть несколько групп, которые увеличивают эффективность соединения, в основном амиды, амины и тетразолы но также метилсульфонильные и трифторметильные группы. Из этих групп карбоксамид имеет наибольшее связывание и обладает такой же свертывающей активностью, что и соединения.[25]
При тестировании на собаках это соединение с группой карбоксамида под названием 13F показало отличный фармакокинетический профиль, низкий клиренс и адекватный период полураспада и объем распространения. Из-за успеха поиска стабилизирующей группы исследование SAR для S1-связывающего фрагмента (п-метоксифенил) было прекращено. В группе связывания S4 было доказано, что аналоги N-метилацетила и лактама обладают очень высокой аффинностью связывания с FXa, показали большую свертываемость и селективность по сравнению с другими протеазами. Ориентация оказалась важной, поскольку N-метилацетил, по сравнению с ацетамид, имел в 300 раз более низкую способность связывания с FXa из-за неблагоприятной планарности вблизи сайта связывания области S4.[25]
Синтез
Ривароксабан
Ривароксабан химически принадлежит к группе н-арилоксазолидиноны. Другие препараты этой группы: линезолид и тедизолид, оба из которых антибиотики. Синтез н-арилоксазолидинонов, начиная с О-силил защищенный этил (2,3-дигидроксипропил) -карбамат был опубликован в 2016 году. В реакции в одном сосуде карбамат циклизируется до 2-оксазолидонового кольца в слабоосновных условиях, в то время как азот оксазолидона одновременно является арилированный к медь -катализация. В частности, для ривароксабана 3-морфолинон заменяет йод в p-позиции бензол звонить медь -катализация. После этого силил защитная группа удаляется, и в результате алкоголь заменяется аминогруппа что тогда ацилированный на последнем этапе.[26]
Промышленный препарат ривароксабана зарегистрирован как патент к Bayer Healthcare в 2005 году.[27] Он начинается с N- (4-аминофенол) -морфолинона, который является алкилированный по оксид пропилена производное, которое также содержит первичный амин, участвующий в фталимид группа защиты. Далее фосген эквивалент добавлен, чтобы сформировать 2-оксазолидон кольцо и фталимид удаляется. Теперь свободный амин можно ацилировать, что приводит к ривароксабану.
Однако, согласно патенту, синтез имеет «различные недостатки в управлении реакцией, которые имеют особенно неблагоприятные последствия для получения». Патент также объясняет другой синтез, начиная с хлоротиофен производное, которое больше подходит для промышленного процесса, но указывает на токсичность растворители или реагенты должны быть удалены из конечного продукта. Следовательно, этот способ не является альтернативой.[27]
Описаны различные другие пути синтеза ривароксабана.[28][29]
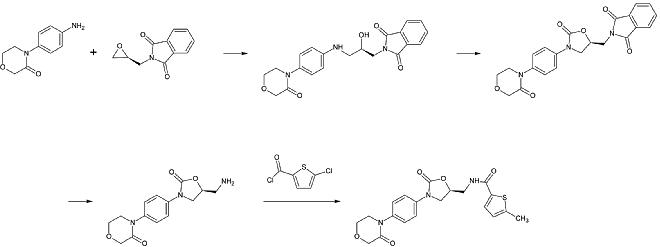
1-й этап: алкилирование первичного ароматического амина
2-й этап: образование 2-оксазолинидонового кольца с использованием эквивалента фосгена
3-й шаг: Удаление группы защиты фталимида
4-й этап: ацилирование первичного амина
Апиксабан
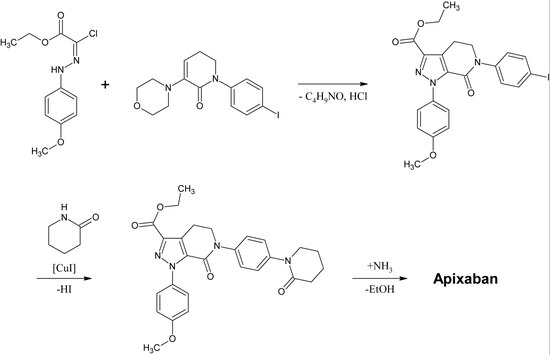
Первый полный синтез апиксабана был опубликован в 2007 году.[30] Ключевым этапом этой реакции является (3 + 2)циклоприсоединение производного п-метоксифенилхлоргидразона и производного п-иодфенилморфолин-дигидропиридина. После следующих устранение из HCl и морфолин, то йод заменяется на 2-пиперидинон к медь -катализация и этилэфир превращается в амид (аминолиз ). Эта реакция была зарегистрирована как патент в 2009 году.[31]
Клиническое использование
Прямые ингибиторы фактора Ха используются в клинической практике, и их использование постоянно увеличивается. Они постепенно захватывают варфарин использование и низкомолекулярные гепарины (НМВ ).[8] Показанием к применению ингибиторов Ха является профилактика глубокие венозные тромбы (ТГВ), что может привести к легочная эмболия. Он также используется для лечения мерцательная аритмия для снижения риска инсульта, вызванного тромбом. Еще одно показание - профилактическое лечение свертывания крови (тромбоз ) из-за атеросклероз. Ривароксабан был первым ингибитором FXa на рынке, за ним последовали апиксабан, эдоксабан и Betrixaban.
| Ривароксабан | Апиксабан | Эдоксабан | Бетриксабан | |
|---|---|---|---|---|
| Имя бренда | Xarelto | Eliquis | Савайса, Ликсиана | Bevyxxa |
| Разработчик и продюсер | Байер | Pfizer | Дайичи Санкё | Portola Pharmaceuticals |
Фармакокинетика
| Ривароксабан | Апиксабан | Эдоксабан | |
|---|---|---|---|
| Метаболизм | CYP3A4 / 5 (основной), CYP2J2 (второстепенный) | CYP3A4 (основной), CYP1A2, 2C8, 2C19, 2J2 (все второстепенные) | CYP34A (основной) |
| Связывание с белками (%) | 92–95 | 87 | 55 |
| Период полураспада (часы) | 5–9 | 6–12 | 5–11 |
| Устранение | Почечный (66%; 36% в неизмененном виде) | Почечные (27%), фекальные | Почечный (35%) |
| Поглощение (Tmax) | 2–4 часа | 3–4 часа | 1-2 часа |
| Распределение (L) | 50 | 21–61 | 107 |
| Почечный клиренс (л / час) | 2.4 | 7.5 | 11 |
Будущие перспективы
Прямые ингибиторы Ха в клинических испытаниях
Ривароксабан, апиксабан, эдоксабан и бетриксабан уже представлены на рынке. По состоянию на октябрь 2016 г. в клинические испытания прошли несколько новых прямых ингибиторов Ха. Это летаксабан от компании Takeda и эрибаксабан от компании Pfizer.[34]
Противоядия
Andexxa из Portola Pharmaceuticals это рекомбинантный белок что дано внутривенно. Он работает как противоядие ко всем прямым и непрямым ингибиторам FXa. Andexxa действует как рецептор-ловушка для ингибиторов Ха.
Рекомендации
- ^ а б «Европейское агентство по лекарственным средствам. 2016. Xarelto». www.ema.europa.eu. Получено 2016-10-03.
- ^ «Европейское агентство по лекарственным средствам. 2016. Eliquis». www.ema.europa.eu. Получено 2016-10-03.
- ^ Бханура, Сангита; Ахлувалия, Каза (01.01.2014). «Новый ингибитор фактора Ха: Апиксабан». Журнал фармакологии и фармакотерапии. 5 (1): 12–4. Дои:10.4103 / 0976-500x.124409. ЧВК 3917159. PMID 24554904.
- ^ Чан, Льюи; Пизано, Микеле (03.10.2016). «Эдоксабан (Савайса): ингибитор фактора Ха». Аптека и терапия. 40 (10): 651–95. ISSN 1052-1372. ЧВК 4606855. PMID 26535021.
- ^ а б Wardrop, D .; Килинг, Д. (2008). «История открытия гепарина и варфарина». Британский журнал гематологии. 141 (6): 757–63. Дои:10.1111 / j.1365-2141.2008.07119.x. PMID 18355382.
- ^ Фрэнсис, CW. (2008). «Варфарин: историческая перспектива». Гематология. 2008: 251. Дои:10.1182 / asheducation-2008.1.251. PMID 19074091.
- ^ а б c Massimo, F .; Маннуччи, П.М. (2016). «Прямые пероральные антикоагулянты и венозная тромбоэмболия». Европейский респираторный обзор. 25 (141): 295–302. Дои:10.1183/16000617.0025-2016. PMID 27581829.
- ^ а б c d е ж грамм час я j k Perzborn, E .; Roehrig, S .; Straub, A .; Кубица, Д .; Миссельвиц, Ф. (2011). «Открытие и разработка ривароксабана, перорального прямого ингибитора фактора Ха». Обзоры природы Drug Discovery. 10 (1): 61–75. Дои:10.1038 / nrd3185. PMID 21164526.
- ^ Бауэр, К. (2013). «Плюсы и минусы новых пероральных антикоауглантов». Гематология. 2013: 464–70. Дои:10.1182 / asheducation-2013.1.464. PMID 24319220.
- ^ а б Фьюри, B; Фьюри, Британская Колумбия (2008). «Механизмы тромбообразования». Медицинский журнал Новой Англии. 359 (9): 938–49. Дои:10.1056 / nejmra0801082. PMID 18753650.
- ^ Davie, E.W .; Fujikawa, K; Кисиэль, W (1991). «Каскад коагуляции: инициирование, поддержание и регулирование». Биохимия. 30 (43): 10363–70. Дои:10.1021 / bi00107a001. PMID 1931959.
- ^ Mackman, N; Tilley, R.E .; Ки, Н. (2007). «Роль внешнего пути свертывания крови в гемостазе и тромбозе». Артериосклероз, тромбоз и биология сосудов. 27 (8): 1687–93. Дои:10.1161 / atvbaha.107.141911. PMID 17556654.
- ^ а б c Lapatto, R .; Кренгель, У .; Schreuder, H.A .; Arkema, A .; де Бур, В .; Kalk, K. H .; Hol, W. G .; Grootenhuis, P. D .; Малдерс, Дж. У. (1 сентября 1997 г.). «Рентгеновская структура антистазина с разрешением 1,9 A и его смоделированный комплекс с фактором свертывания крови Ха». Журнал EMBO. 16 (17): 5151–61. Дои:10.1093 / emboj / 16.17.5151. ISSN 0261-4189. ЧВК 1170148. PMID 9311976.
- ^ Шульц, Лорен Д .; Маркус, Генри З .; Хофманн, Кэтрин Дж .; Монтгомери, Донна Л .; Данвидди, Кристофер Т .; Kniskern, Peter J .; Фридман, Роберт Б .; Эллис, Рональд У .; Туите, Майкл Ф. (1 июня 1994). «Использование молекулярной генетики для улучшения производства рекомбинантных белков дрожжевыми Saccharomyces cerevisiae». Летопись Нью-Йоркской академии наук. 721 (1): 148–57. Дои:10.1111 / j.1749-6632.1994.tb47387.x. ISSN 1749-6632. PMID 8010665.
- ^ Нагахара, Такаясу; Ёкояма, Юкио; Инамура, Казуэ; Катакура, Син-ичи; Комория, Сатоши; Ямагути, Хитоши; Хара, Цуёси; Ивамото, Масахиро (1 апреля 1994 г.). «Производные двухосновной (амидиноарил) пропановой кислоты как новые ингибиторы фактора свертывания крови Ха». Журнал медицинской химии. 37 (8): 1200–07. Дои:10.1021 / jm00034a018. ISSN 0022-2623. PMID 8164262. S2CID 19381209.
- ^ Сато, Кадзуо; Кавасаки, Томихиса; Таниучи, Юта; Хираяма, Фукуши; Кошио, Хироюки; Мацумото, Юдзо (1997-11-27). «YM-60828, новый ингибитор фактора Ха: разделение его антитромботических эффектов и увеличения времени кровотечения». Европейский журнал фармакологии. 339 (2–3): 141–46. Дои:10.1016 / S0014-2999 (97) 01389-7. PMID 9473127.
- ^ «Краткое обоснование решения (SBD) для PrXARELTO». Министерство здравоохранения Канады. 2009-02-13. Архивировано из оригинал на 2016-10-09. Получено 2016-10-03.
- ^ а б c d Нар, Герберт (2012). «Роль структурной информации в открытии прямых ингибиторов тромбина и фактора Ха». Тенденции в фармакологических науках. 33 (5): 279–88. Дои:10.1016 / j.tips.2012.03.004. PMID 22503439.
- ^ Брандштеттер, Блэнд (1996). «Рентгеновская структура фактора Xa свертывания, ингибируемого активным сайтом». Журнал биологической химии. 271 (47): 29988–92. Дои:10.1074 / jbc.271.47.29988. PMID 8939944.
- ^ а б c Стейнберг, Бенджамин А. (2014). «Структурно-функциональные отношения ингибиторов фактора Ха: значение для практикующего врача». Журнал тромбоза и тромболизиса. 37 (2): 234–41. Дои:10.1007 / s11239-013-0991-z. PMID 23996500.
- ^ Беккер, Ричард С .; Александр, Джон; Дайк, Кристофер К .; Харрингтон, Роберт А. (2004-12-01). «Разработка DX-9065a, нового прямого антагониста фактора Ха, при сердечно-сосудистых заболеваниях». Тромбоз и гемостаз. 92 (6): 1182–93. Дои:10.1160 / TH04-05-0289. ISSN 0340-6245. PMID 15583722. S2CID 953689.
- ^ а б Katakura, S .; Hara, T .; Nagahara, T .; Kunitada, S .; Ивамото, М. (1995-05-01). «Молекулярная модель взаимодействия между фактором Ха и DX-9065a, новым ингибитором фактора Ха: вклад ацетимидоилпирролидиновой части ингибитора в эффективность и селективность сериновых протеаз». Европейский журнал медицинской химии. 30 (5): 387–94. Дои:10.1016/0223-5234(96)88248-1.
- ^ Пинто, Орват, Кох, Дональд Дж. П. Майкл Дж. Стефани (2007). "Открытие 1- (4-Метоксифенил) -7-оксо-6- (4- (2-оксопиперидин-1-ил) фенил) -4,5,6,7-тетрагидро-1H-пиразоло [3,4- c] пиридин-3-карбоксамид (Апиксабан, BMS-562247), высокоэффективный, селективный, эффективный и перорально биодоступный ингибитор фактора свертывания крови Ха ». Журнал медицинской химии. 50 (22): 5339–56. Дои:10.1021 / jm070245n. PMID 17914785.CS1 maint: несколько имен: список авторов (связь)
- ^ Рериг, Сюзанна (2005). "Открытие нового антитромботического агента 5-Хлор-N - ({(5 S) -2-оксо-3- [4- (3-оксоморфолин-4-ил) фенил] -1,3-оксазолидин-5-ил ". Журнал медицинской химии. 48 (19): 5900–5908. Дои:10.1021 / jm050101d. PMID 16161994.
- ^ а б Пинто, Д. Дж .; Орват, М. Дж .; Koch, S .; Росси, К. А .; Александр, Р. С .; Смоллвуд, А .; Лам, П. Я. (2007). "Открытие 1- (4-метоксифенил) -7-оксо-6- (4- (2-оксопиперидин-1-ил) фенил) -4,5,6,7-тетрагидро-1H-пиразоло [3,4- в] пиридин-3-карбоксамид (апиксабан, BMS-562247), высокоэффективный, селективный, эффективный и биодоступный при пероральном приеме ингибитор фактора свертывания крови Ха ». J Med Chem. 50 (22): 5339–56. Дои:10.1021 / jm070245n. PMID 17914785.
- ^ Махи, Уильям; Leitch, Jamie A .; Фрост, Кристофер Г. (2016-03-01). «Катализируемая медью сборка N-арилоксазолидинонов: синтез линезолида, тедизолида и ривароксабана». Европейский журнал органической химии. 2016 (7): 1305–13. Дои:10.1002 / ejoc.201600033. ISSN 1099-0690.
- ^ а б c Патент США US7351823, Матиас Берве, Кристиан Томас, Иоахим Резе, Дирк Гротйоханн, «Процесс подготовки», опубликовано 2008-04-D01, выпущено 10 января 2005 г.
- ^ Ли, Чао; Лю, Иншуай; Чжан, Юнцзюнь; Чжан, Синсянь (01.07.2011). «Подход к антикоагулянтному агенту ривароксабану через циклоприсоединение изоцианат-оксиран, продвигаемое MgI2-эфиром». Журнал химических исследований. 35 (7): 400–01. Дои:10.3184 / 174751911X13098778358582.
- ^ Юань, Цзяньюн; Лю, Кай; Ли, Лун; Юань, Юн; Лю, Сюэлей; Ли, Янву (18 сентября 2014 г.). «Новый синтез оксазолидинонового антитромботического агента ривароксабана». Молекулы. 19 (9): 14999–15004. Дои:10.3390 / молекулы190914999. ЧВК 6271174. PMID 25237754.
- ^ а б Пинто, Дональд Дж. П .; Орват, Майкл Дж .; Кох, Стефани; Росси, Карен А .; Александр, Ричард С .; Смоллвуд, Анджела; Wong, Pancras C .; Рендина, Алан Р .; Люетген, Джозеф М. (2007-11-01). "Открытие 1- (4-Метоксифенил) -7-оксо-6- (4- (2-оксопиперидин-1-ил) фенил) -4,5,6,7-тетрагидро-1H-пиразоло [3,4- c] пиридин-3-карбоксамид (Апиксабан, BMS-562247), высокоэффективный, селективный, эффективный и перорально биодоступный ингибитор фактора свертывания крови Ха ». Журнал медицинской химии. 50 (22): 5339–56. Дои:10.1021 / jm070245n. ISSN 0022-2623. PMID 17914785.
- ^ Патент США US 20100130543 A1, Thomas G. Gant, Manoucherhr M. Shahbaz, "Пиразолкарбоксамидные ингибиторы фактора xa", опубликовано 27 мая 2010 г., опубликовано 14 сентября 2009 г.
- ^ Frost, C .; Песня, Y .; Barret, Y.C .; Wang, J .; Персли, Дж. (2014). «Рандомизированное прямое сравнение фармакокинетики и фармакодинамики апиксабана и ривароксабана». Клиническая фармакология. 6: 179–87. Дои:10.2147 / CPAA.S61131. ЧВК 4235474. PMID 25419161.
- ^ Parasrampuriam, D.A .; Труитт, К. (2016). «Фармакокинетика и фармакодинамика эдоксабана, перорального антикоагулянта, не являющегося антагонистом витамина К, который ингибирует фактор свертывания крови Ха». Клиническая фармакокинетика. 55 (6): 641–55. Дои:10.1007 / s40262-015-0342-7. ЧВК 4875962. PMID 26620048.
- ^ Аренс, я; Karlheinz, P .; Губа, GYH .; Боде, К. (июнь 2012 г.). «Разработка и клиническое применение новых пероральных антикоагулянтов. Часть II. Лекарства, находящиеся в стадии клинических исследований». Открытие Мейдицин. 13 (73): 445–50. PMID 22742650.