Список первичных иммунодефицитов - List of primary immunodeficiencies
Это список первичные иммунодефициты (PID), которые иммунная недостаточность которые не являются вторичными по отношению к другому состоянию.
В Международный союз иммунологических обществ распознает девять классов первичных иммунодефицитов, всего около 350 состояний.[1] В обновлении руководства по классификации 2014 г. добавлена 9-я категория и 30 новых дефектов генов по сравнению с предыдущей версией 2009 г.[2][3] Самая последняя классификация была выпущена в 2017 году. Число выявленных состояний продолжает расти с течением времени по мере проведения дополнительных исследований.
Воздействие первичных иммунодефицитов варьируется от легкого до тяжелого в зависимости от состояния.[4]
Комбинированные Т- и В-клеточные иммунодефициты
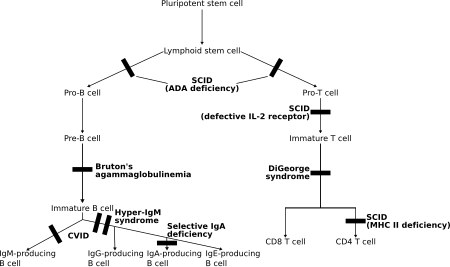
При этих расстройствах оба Т-лимфоциты и часто В-лимфоциты, регуляторы адаптивного иммунитета, дисфункциональны или уменьшены в количестве. Основными членами являются различные типы тяжелый комбинированный иммунодефицит (ТКИД).[5]
- T- / B + SCID (преимущественно Т-лимфоциты отсутствуют):
- дефицит γc
- Дефицит JAK3
- Рецептор интерлейкина-7-α недостаток
- CD45 недостаток
- CD3δ, CD3ε или CD3ζ недостаток
- Коронин-1А недостаток
- LAT (ген) недостаток
- T- / B- SCID (отсутствуют как Т-, так и В-клетки)
- ТРЯПКА 1/2 недостаток
- DCLRE1C (Артемида) дефицит
- XLF (белок) /Дефицит цернунноса
- ДНК PKcs недостаток
- ДНК-лигаза дефицит IV типа
- аденозиндезаминаза (ADA) дефицит
- ретикулярная дисгенезия
- Синдром Оменна
- Лиганд CD40 недостаток
- CD40 недостаток
- CD3γ недостаток
- CD8 недостаток
- ICOS недостаток
- ZAP70 недостаток
- Дефицит канала Ca ++
- MHC класс I дефицит (с мутациями в TAP1, TAP2, ТАПБП, или же B2M )
- MHC класс II дефицит (с мутациями в CIITA, RFXANK, RFX5, или же RFXAP )
- Дефицит CD25
- Дефицит CD27
- STAT5b дефицит
- Дефицит ITK
- SH2D1A дефицит (XLP1)
- Дефицит MAGT1
- DOCK2 недостаток
- DOCK8 дефицит
- Дефицит RhoH
- Синдром активированного дельта PI3K
- MALT1 недостаток
- BCL10 недостаток
- BCL11B недостаток
- CARD11 недостаток
- Дефицит MST1
- TCRα дефицит
- Дефицит LCK
- Ил-21 недостаток
- Дефицит IL-21R
- UNC119 дефицит
- Дефицит НИК
- Дефицит OX40
- IKBKB дефицит
- TFRC недостаток
- Моэсин недостаток
- RELB недостаток
- Гипоплазия хрящевых волос
- Дефицит LRBA
Преимущественно дефицит антител
В начальной дефицит антител, один или несколько изотипов иммуноглобулин уменьшаются или не работают должным образом. Эти белки, генерируемые плазматические клетки, обычно связываются с патогенами, направляя их на уничтожение.[5]
- Отсутствие B-клеток, что приводит к серьезному снижению всех типов антител: Х-сцепленная агаммаглобулинемия (btk дефицит, или Bruton агаммаглобулинемия), μ -Тяжелая цепь дефицит, l 5 дефицит, Igα дефицит, BLNK дефицит, тимома с иммунодефицитом
- Уровень B-клеток низкий, но присутствует или нормальный, но со снижением 2 или более изотипов (обычно IgG и IgA, иногда IgM): общий вариабельный иммунодефицит (CVID), CD19 дефицит, ТАСИ (TNFRSF13B) дефицит, Рецептор BAFF дефицит.
- Нормальное количество В-клеток с пониженным IgG и IgA и увеличился IgM: Синдромы гипер-IgM
- Нормальное количество В-клеток с дефицитом изотипа или легкой цепи: тяжелая цепь удаления, каппа цепь дефицит, изолированный дефицит подкласса IgG, IgA с дефицитом подкласса IgG, селективный дефицит иммуноглобулина А
- Специфическая недостаточность антител к специфическим антигенам с нормальными B-клетками и нормальными концентрациями Ig
- Преходящая гипогаммаглобулинемия младенческого возраста (THI)
Другой четко выраженный синдром иммунодефицита
Ряд синдромов не подлежит официальной классификации, но в остальном распознается по определенным клиническим или иммунологическим признакам.[5]
- Иммунодефицит с тромбоцитопенией
- Ремонт ДНК дефекты, не вызывающие изолированного SCID:
- Атаксия-телеангиэктазия
- Атаксиоподобный синдром
- Синдром перелома Неймегена
- Синдром Блума
- Синдром иммунодефицита - центромерной нестабильности - лицевых аномалий (ICF1, 2, 3 и 4)
- Дефицит PMS2
- ЗАГАДКА синдром (Дефицит RNF168)
- Дефицит MCM4
- Синдром FILS (СТОЛБ дефицит)
- ПОЛЮС 2 недостаток
- LIG1 недостаток
- NSMCE3 недостаток
- Дефицит гебо
- GINS1 недостаток
- Синдром ДиДжорджи (когда связан с тимус дефекты)
- TBX1 недостаток
- ЗАРЯДНЫЙ синдром (CHD7 дефицит или SEMA3E дефицит)
- Крылатая спираль /FOXN1 недостаток
- Делеция 10p13-p14 хромосомы
- Иммуно-костные дисплазии (аномальное развитие скелета с проблемами иммунитета):
- Гипоплазия волос и хрящей
- Синдром Шимке
- MYSM1 недостаток
- MOPD1 недостаток
- EXTL3 недостаток
- Синдромы гипер-IgE
- Синдром работы (STAT3 дефицит)
- Синдром Комеля-Нетертона
- PGM3 недостаток
- Гипогидротическая эктодермальная дисплазия
- Дефекты кальциевых каналов
- ORAI1 недостаток
- Дефицит STIM1
- Транскобаламин 2 недостаток
- Иммунодефицит с множественными атрезиями кишечника (TTC7A дефицит)
- Веноокклюзионная болезнь печени с иммунодефицитом (VODI)
- Синдром Вичи
- Дефицит пуриновой нуклеозидфосфорилазы (PNP)
- AR-DKC (аутосомно-доминантный дискератоз врожденный)
- Синдром Германского-Пудлака тип 2
- Хронический кожно-слизистый кандидоз
- HOIL1 недостаток
- HOIP недостаток
- XL-врожденный дискератоз (Синдром Хойераала-Хрейдарссона )
- Синдром лимфангиэктазии-лимфедемы Хеннекама
- Синдром Кабуки
- Дефицит MTHFD1
- STAT5b дефицит
- Дефицит ИКАРОС
Заболевания иммунной дисрегуляции
В определенных условиях преобладающей проблемой является регуляция, а не внутренняя активность частей иммунной системы.[5]
- Иммунодефицит с гипопигментацией или альбинизм: Синдром Чедиака – Хигаши, Синдром Грищелли тип 2
- Семейный гемофагоцитарный лимфогистиоцитоз: перфорин дефицит, UNC13D дефицит, синтаксин 11 недостаток
- Х-сцепленный лимфопролиферативный синдром
- Синдромы с аутоиммунитетом:
- (а) Аутоиммунный лимфопролиферативный синдром: тип 1а (CD95 дефекты), тип 1б (Fas лиганд дефекты), тип 2а (CASP10 дефекты), тип 2б (CASP8 дефекты)
- (б) ПРИНЯТО (аутоиммунная полиэндокринопатия с кандидозом и эктодермальной дистрофией)
- (c) IPEX (иммунодисрегуляция, полиэндокринопатия, энтеропатия, Х-сцепленный синдром)
- (г) Дефицит CD25
Врожденные дефекты количества, функции фагоцитов или того и другого
Фагоциты это клетки, которые поглощают и поглощают патогены (фагоцитоз ), и уничтожить их химикатами. Моноциты /макрофаги а также гранулоциты способны на этот процесс. В определенных условиях либо уменьшается количество фагоцитов, либо их функциональная способность нарушается.[5]
- Тяжелая врожденная нейтропения: из-за ELA2 дефицит (с миелодисплазия )
- Тяжелая врожденная нейтропения: из-за GFI1 недостаточность (при Т / В лимфопении)
- Эластаза недостаток
- Синдром Костмана (HAX1 дефицит)
- Нейтропения при пороках сердца и мочеполовой системы
- Болезнь накопления гликогена типа 1b
- Синдром Коэна
- Синдром Клерикуцио
- Циклическая нейтропения
- Х-сцепленная нейтропения / миелодисплазия
- Недостаток P14
- HYOU1 недостаток
- JAGN1 недостаток
- SMARCD2 недостаток
- 3-метилглутаконовая ацидурия
- Нарушение адгезии лейкоцитов 1 типа
- Дефицит адгезии лейкоцитов тип 2
- Дефицит адгезии лейкоцитов тип 3
- RAC2 дефицит (Синдром нейтрофильного иммунодефицита )
- Бета-актин недостаток
- G-CSF-рецептор недостаток
- Локализованный ювенильный пародонтит
- Синдром Папийона-Лефевра
- Специфический дефицит гранул
- Синдром Швахмана-Даймонда
- WDR1 недостаток
- Кистозный фиброз
- Хроническая гранулематозная болезнь: Х-сцепленный или аутосомный (CYBA, NCF1, NCF2, NCF4 )
- Ил-12 и Ил-23 дефицит β1 цепи
- Ил-12п40 недостаток
- Дефицит глюкозо-6-фосфатдегидрогеназы 1 класс
- Рецептор интерферона γ 1 недостаток
- Рецептор интерферона γ 2 недостаток
- STAT1 недостаток
- MKL1 недостаток
- ОБЪЯВЛЕНИЕ гипер-IgE
- AR гипер-IgE
- Легочный альвеолярный протеиноз
- Синдром MonoMac (GATA2 дефицит )
Дефекты врожденного иммунитета
Несколько редких состояний возникают из-за дефектов в врожденная иммунная система, который является основной линией защиты, независимой от более продвинутых систем, связанных с лимфоцитами. Многие из этих состояний связаны с проблемами кожи.[5]
- Рецептор интерлейкина 12, бета 1 недостаток
- Ил-12п40 недостаток
- Гамма-рецептор интерферона 1 недостаток
- Гамма-рецептор интерферона 2 недостаток
- Tyk2 дефицит
- JAK1 Потеря функции
- ISG15 недостаток
- RORc недостаток
- STAT1 дефицит, мутация увеличения функции
- STAT2 недостаток
- IRF7 недостаток
- CD16 недостаток
- IRF8 недостаток
- IFNAR2 недостаток
- Недостатки пути TLR
- MDA5 недостаток
- Бородавчатая эпидермодисплазия
- Синдром WHIM (бородавки, гипогаммаглобулинемия, инфекции, миелокатексис)
- EVER1 и EVER2 недостаток
- Энцефалит, вызванный вирусом простого герпеса
- CARD9 недостаток
- Хронический кожно-слизистый кандидоз
- Трипаносомоз
- RPSA дефицит с врожденная аспления
- HMOX дефицит с врожденной аспленией
- CLCN7 дефицит с остеопороз
- OSTM1 дефицит при остеопорозе
- Гнойный гидраденит
Аутовоспалительные расстройства
Большинство аутовоспалительных заболеваний не предрасполагают к инфекциям, а приводят к чрезмерному воспалению. Многие проявляют себя как синдромы периодической лихорадки. Они могут напрямую поражать различные органы, а также предрасполагать к долгосрочным повреждениям (например, приводя к амилоид осаждение).[5]
- Семейная средиземноморская лихорадка
- Синдром Айкарди – Гутьера с TREX1, SAMHD1 или же IFIH1 мутации
- Спондилоэнхондро-дисплазия с нарушением иммунной регуляции (ACP5 мутация)
- Васкулопатия, связанная с СИНГ, с началом в младенчестве
- Х-сцепленное ретикулярное пигментное расстройство
- USP18 недостаток
- СВЕЧА (Хронический атипичный нейтрофильный дерматит с липодистрофией)
- Синдром Синглтона-Мертена
- Периодический синдром, связанный с рецептором TNF (ЛОВУШКИ)
- Синдром гипер-IgD (Дефицит мевалонаткиназы )
- CIAS1 -связанные заболевания:
- NLRP1 недостаток
- Синдром ПАПА (гнойный стерильный артрит, гангренозная пиодермия, акне)
- ADAM17 недостаток
- Синдром Блау
- Синдром маджида (Хронический рецидивирующий мультифокальный остеомиелит и врожденная дизеритропоэтическая анемия)
- ДИРА (дефицит антагониста рецептора ИЛ-1)
- ДИТРА (дефицит антагониста рецептора IL-36)
- CARD14-опосредованный псориаз (ЛАГЕРЯ)
- Херувизм
- Дефект COPA
- Отулипения / ORAS
Дополнять недостатки
В система комплемента является частью врожденной, а также адаптивной иммунной системы; это группа циркулирующих белков, которые могут связывать патогены и образовывать комплекс мембранной атаки. Дополнять недостатки являются результатом недостатка любого из этих белков. Они могут быть предрасположены к инфекциям, а также к аутоиммунным заболеваниям.[5]
- Дефицит C1q (волчаночный синдром, ревматоидная болезнь, инфекции)
- C1r дефицит (то же самое)
- Дефицит C1s
- Дефицит C4 (волчаночный синдром)
- Дефицит C2 (волчаночный синдром, васкулит, полимиозит, гнойные инфекции )
- Дефицит C3 (повторяющийся гнойные инфекции )
- Дефицит C5 (Нейссериальные инфекции, СКВ)
- Дефицит C6 (то же самое)
- Дефицит C7 (то же самое, васкулит)
- C8a дефицит
- Дефицит C8b
- Дефицит C9 (Нейссериальные инфекции)
- Дефицит С1-ингибитора (наследственный ангионевротический отек)
- Недостаток фактора I (гнойные инфекции )
- Недостаток фактора H (гемолитико-уремический синдром, мембранопролиферативный гломерулонефрит )
- Дефицит фактора D (Нейссериальные инфекции)
- Дефицит пропердина (Нейссериальные инфекции)
- Дефицит MBP (гнойные инфекции )
- MASP2 дефицит
- Дефицит рецептора 3 комплемента
- Дефицит белка мембранного кофактора (CD46)
- Дефицит ингибитора комплекса мембранной атаки (CD59)
- Пароксизмальная ночная гемоглобинурия
- Дефицит фиколина 3
- Дефицит пропердина
- Недостаток фактора I
- Недостаток фактора H
- Дефицит тромбомодулина
- Болезнь ЧАПЕЛЛ
Фенокопия первичного иммунодефицита
- Аутоиммунный лимфопролиферативный синдром
- Аутоиммунное лейкопролиферативное заболевание, связанное с РАС
- Большой гранулярный лимфоцитоз
- Атипичный гемолитико-уремический синдром
- Хороший синдром
Рекомендации
- ^ Бусфиха, Азиз; Джеддейн, Лейла; Пикард, Капуцин; Айлал, Фатима; Бобби Гаспар, H .; Аль-Герц, Валид; Шатила, Талал; Ворона, Яник Дж. (2018). «Фенотипическая классификация первичных иммунодефицитов IUIS 2017 г.». Журнал клинической иммунологии. 38 (1): 129–143. Дои:10.1007 / s10875-017-0465-8. ISSN 0271-9142. ЧВК 5742599. PMID 29226301.
- ^ Валид аль-Герц; Азиз Бусфиха; Жан-Лоран Казанова; и другие. (2014). «Заболевания, связанные с первичным иммунодефицитом: обновленная информация о классификации Комитета экспертов по первичному иммунодефициту Международного союза иммунологических обществ» (PDF). Границы иммунологии. 5 (162): 1–33. Дои:10.3389 / fimmu.2014.00162. ЧВК 4001072. PMID 24795713.
- ^ Notarangelo L, Casanova JL, Conley ME, et al. (2006). «Заболевания, связанные с первичным иммунодефицитом: обновленная информация о заседании Комитета по классификации первичных иммунодефицитных заболеваний Международного союза иммунологических обществ в Будапеште, 2005 г.». J. Allergy Clin. Иммунол. 117 (4): 883–96. Дои:10.1016 / j.jaci.2005.12.1347. PMID 16680902.
- ^ «Общий вариабельный иммунодефицит». NORD (Национальная организация по редким заболеваниям). Получено 5 марта 2019.
- ^ а б c d е ж грамм час Notarangelo LD, Fischer A, Geha RS, et al. (Декабрь 2009 г.). «Первичные иммунодефициты: обновленная информация за 2009 год: Комитет экспертов Международного союза иммунологических обществ (IUIS) по первичным иммунодефицитам (PID)». J. Allergy Clin. Иммунол. 124 (6): 1161–78. Дои:10.1016 / j.jaci.2009.10.013. ЧВК 2797319. PMID 20004777.