Почечный канальцевый ацидоз - Renal tubular acidosis
| Почечный канальцевый ацидоз | |
|---|---|
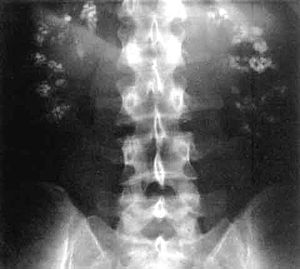 | |
| Существенный двусторонний нефрокальциноз (кальциноз почек) на лобной рентгеновский снимок (рентгеноконтрастность (белый цвет) в правом верхнем и левом верхнем квадранте изображения), как видно при ацидозе дистальных почечных канальцев | |
| Специальность | Нефрология |
Почечный канальцевый ацидоз (RTA) - это заболевание, при котором происходит накопление кислота в организме из-за отказа почки должным образом подкислять то моча.[1] В почечная физиология, когда кровь является фильтрованный почками фильтрат проходит через трубочки из нефрон, позволяющий обменивать соли, кислотные эквиваленты и другие растворенные вещества прежде, чем он стечет в мочевой пузырь в качестве моча. В Метаболический ацидоз результат ДТП может быть вызван несоблюдением впитывать достаточный бикарбонат ионы (которые щелочной ) из фильтрата в ранней части нефрона ( проксимальный каналец ) или недостаточным секреция из ионы водорода (которые являются кислыми) в последние части нефрона ( дистальный каналец ). Хотя метаболический ацидоз также встречается у людей с хроническая болезнь почек, термин RTA предназначен для людей с плохим закислением мочи в нормально функционирующих почках. Существует несколько различных типов ДТП, каждый из которых имеет разные синдромы и разные причины. RTA обычно является случайной находкой, основанной на обычных анализах крови, которые показывают ненормальные результаты. Клинически пациенты могут иметь нечеткие симптомы, такие как обезвоживание, изменение психического статуса или задержка роста у подростков. [2]
Слово ацидоз относится к тенденции RTA вызывать превышение кислота, что снижает pH. Когда pH крови ниже нормы (7,35), это называется ацидемия. Метаболический ацидоз, вызванный RTA, представляет собой ацидоз с нормальной анионной щелью.
Типы
Обзор типов 1, 2 и 4 представлен ниже (тип 3 обычно исключается из современных классификаций):
| Тип | Тип 1 | Тип 2 | Тип 4 |
|---|---|---|---|
| Место расположения | Сборные канальцы, дистальные канальцы | Проксимальные канальцы | Надпочечники |
| Ацидемия | Да (очень серьезно) | да | Мягкий при наличии |
| Калий | Гипокалиемия | Гипокалиемия | Гиперкалиемия |
| Патофизиология | Отказ α интеркалированные клетки секретировать H+ и вернуть K+ | Отказ клетки проксимальных канальцев впитывать ЧАСCО− 3 | Дефицит альдостерон, или сопротивление его эффектам, (гипоальдостеронизм или же псевдогипоальдостеронизм ) |
Тип 1: дистальный
Дистальный RTA (dRTA) - это классическая форма RTA, описанная первой. Дистальный RTA характеризуется нарушением секреции H + в просвет нефрона альфа-интеркалированным клетки из медуллярный собирательный проток из дистальный нефрон.
Этот недостаток секреции кислоты может быть вызван рядом причин и приводит к неспособности подкислять мочу до pH менее 5,3. Поскольку почечная экскреция является основным средством устранения ЧАС+
от тела, следовательно, есть тенденция к ацидемия. Нет возможности выводить H+ пока K+
не могут быть восстановлены клеткой, что приводит к ацидемии (поскольку ЧАС+
накапливается в организме) и гипокалиемия (как K+
не может быть повторно поглощен альфа-клеткой).
Это приводит к клиническим особенностям dRTA;[1] Другими словами, апикальный H + / K + -антипортер интеркалированных клеток нефункционален, что приводит к задержке протонов и экскреции калия. Поскольку кальций-фосфатные камни демонстрируют склонность к отложению при более высоких pH (щелочных), в веществе почек камни образуются с двух сторон; этого не происходит в других типах RTA.
- Нормальный анионная щель Метаболический ацидоз / ацидемия
- Гипокалиемия, Гипокальциемия, Гиперхлоремия
- Мочевой камень образование (связанное с щелочной мочой, гиперкальциурия, и низкий уровень цитрата мочи).[3]
- Нефрокальциноз (отложение кальций в веществе почки)
- Кость деминерализация (вызывающая рахит у детей и остеомаляция у взрослых
- Дефицит роста
- Медуллярные кисты
- Нейросенсорная тугоухость
- Наследственная гемолитическая анемия
Дистальный RTA также был связан с определенными генетическими мутациями, которые изменятся, когда болезнь проявится в жизни пациента. Пациенты с мутациями в ATP6V1B1 и ATP6V0A4 появятся симптомы в течение первого года жизни, а у пациентов с мутацией SLC4A1 отложили начало около 10 лет.[4] Электролитный дисбаланс остается прежним, а в тяжелых случаях симптомы могут прогрессировать до аминоацидурия и гипераммониемия. [5]. В большой азиатской серии дистального почечного тубулярного ацидоза при синдроме Шегрена поздняя диагностика является правилом, несмотря на явный гипокалиемический периодический паралич у подавляющего большинства из них.[6]
dRTA является наиболее распространенной формой RTA, диагностируемой в западных странах, и может быть классифицирован как наследственный (первичный) или приобретенный (вторичный). Первичная ДТА обычно возникает в результате системных и аутоиммунных заболеваний.[7] или воздействие лекарств и токсинов у взрослых, тогда как педиатрическая ДПТ является результатом генетических дефектов белков, которые способствуют подкислению мочи в дистальных канальцах. Наследственная dRTA обычно проявляется как неспособность к развитию в течение первых нескольких месяцев жизни. Другие общие клинические проявления у детей включают различные симптомы со стороны желудочно-кишечного тракта и мочевыводящих путей, включая полиурию, полидипсию, запор, диарею, приступы обезвоживания и снижение аппетита.[8]
Тип 2: проксимальный

Проксимальный RTA (pRTA) вызван неисправностью проксимальный трубчатый клетки реабсорбируют фильтрованный бикарбонат из мочи, что приводит к бикарбонат расточительство и последующие ацидемия. Реабсорбция бикарбоната обычно составляет 80-90% в проксимальном канальце, и отказ этого процесса приводит к снижению системного буфера и Метаболический ацидоз.[9] Дистальные интеркалированные клетки функционируют нормально, поэтому ацидемия менее серьезна, чем dRTA, и интеркалированные альфа-клетки могут продуцировать H+ для подкисления мочи до pH менее 5,3.[10] pRTA также имеет несколько причин и иногда может присутствовать в виде одиночного дефекта, но обычно связана с более общей дисфункцией проксимальных канальцевых клеток, называемой Синдром Фанкони, в котором также есть фосфатурия, глюкозурия, аминоацидурия, урикозурия и тубулярная протеинурия.
Основным признаком синдрома Фанкони является деминерализация костей (остеомаляция или же рахит ) из-за потери фосфата.
Тип 3: комбинированный проксимальный и дистальный
У некоторых пациентов RTA имеет общие черты как dRTA, так и pRTA. Эта редкая картина наблюдалась в 1960-х и 1970-х годах как временное явление у младенцев и детей с dRTA (возможно, в связи с некоторыми экзогенными факторами, такими как высокое потребление соли) и больше не наблюдается.[11] Эта форма ДТП также называется ДТП несовершеннолетних.[12]
Комбинированный dRTA и pRTA также наблюдается в результате наследственной карбоангидраза II дефицит. Мутации в гене, кодирующем этот фермент, вызывают аутосомно-рецессивный синдром остеопетроз, почечный тубулярный ацидоз, мозговой кальцификация, и умственная отсталость.[13][14][15] Это очень редко, и зарегистрированы случаи со всего мира, из которых около 70% приходится на Магриб регион Северной Африки, возможно, из-за высокой распространенности кровное родство там.[16]Проблемы с почками лечат, как описано выше. Не существует лечения остеопетроза или церебральной кальцификации.
Тип 3 редко обсуждается.[17] Большинство сравнений RTA ограничиваются сравнением типов 1, 2 и 4.
Тип 4: абсолютный гипоальдостеронизм или нечувствительность к альдостерону

RTA 4-го типа на самом деле вообще не является канальцевым заболеванием и не имеет клинического синдрома, аналогичного другим типам RTA, описанным выше. Он был включен в классификацию почечных канальцевых ацидозов, поскольку связан с умеренным (нормальным анионным промежутком) метаболическим ацидозом из-за физиологический уменьшение проксимального трубчатого аммоний экскреция (нарушение аммиагенеза), вторичная по отношению к гипоальдостеронизм, и приводит к снижению буферной способности мочи. Его кардинальная особенность - гиперкалиемия, и измеренное закисление мочи является нормальным, поэтому его часто называют гиперкалиемической ПТА или тубулярной гиперкалиемией.[17]
Причины включают:
- Дефицит альдостерона (гипоальдостеронизм ): Первичная или гипоренемическая (включая диабетическую нефропатию)
- Альдостерон сопротивление
- Наркотики: НПВП, Ингибиторы АПФ и БРА, Эплеренон, Спиронолактон, Триметоприм, Пентамидин
- Псевдогипоальдостеронизм
История
Почечный канальцевый ацидоз был впервые описан в 1935 г. Лайтвудом и в 1936 г. Батлером и соавт. у детей.[18][19] Baines et al. впервые описал его у взрослых в 1945 году.[20]
Дональд Л. Льюис постулировал характер Крошечный Тим, из Рождественская песня, страдал почечным канальцевым ацидозом.[21]
Исследователи опубликовали PLOS ONE в 2009 году предположил, что печально пораженные Карл II Испании могли страдать почечным канальцевым ацидозом в сочетании с комбинированный дефицит гормона гипофиза.[22]
Смотрите также
- Карл II Испании, который предположительно пострадал от dRTA
- Гиперхлоремический ацидоз
- Гипокалиемический ацидоз
- Синдром Лайтвуда – Олбрайта
Рекомендации
- ^ а б Лэйнг К.М., Той А.М., Капассо Дж., Анвин Р.Дж. (июнь 2005 г.). «Почечный канальцевый ацидоз: развитие нашего понимания молекулярных основ». Международный журнал биохимии и клеточной биологии. 37 (6): 1151–61. Дои:10.1016 / j.biocel.2005.01.002. PMID 15778079.
- ^ Яксли Дж., Пирроне С. (21 декабря 2016 г.). «Обзор диагностической оценки почечного тубулярного ацидоза». Журнал Окснера. 16 (4): 525–530. ЧВК 5158160. PMID 27999512.
- ^ Buckalew В.М. (март 1989 г.). «Нефролитиаз при почечном канальцевом ацидозе». Журнал урологии. 141 (3, п. 2): 731–7. Дои:10.1016 / S0022-5347 (17) 40997-9. PMID 2645431.
- ^ Алонсо-Варела М., Хиль-Пенья Х., Кото Е., Гомес Дж., Родригес Дж., Родригес-Рубио Е., Сантос Ф. (сентябрь 2018 г.). «Дистальный почечный канальцевый ацидоз. Клинические проявления у пациентов с различными мутациями основного гена». Детская нефрология. 33 (9): 1523–1529. Дои:10.1007 / s00467-018-3965-8. HDL:10651/48680. PMID 29725771.
- ^ Clericetti CM, Milani GP, Lava SA, Bianchetti MG, Simonetti GD, Giannini O (март 2018 г.). «Гипераммонемия, связанная с ацидозом дистальных почечных канальцев или инфекцией мочевыводящих путей: систематический обзор». Детская нефрология. 33 (3): 485–491. Дои:10.1007 / s00467-017-3829-7. PMID 29134448.
- ^ Сандхья, Пулукул; Данда, Дебашиш; Раджаратнам, Саймон; Томас, Нихал (2014-12-19). "Сьегрен, почечный тубулярный ацидоз и остеомаляция - азиатская индийская серия". Открытый ревматологический журнал. 8: 103–109. Дои:10.2174/1874312901408010103. ISSN 1874-3129. ЧВК 4286932. PMID 25584094.
- ^ Сантос Ф., Ордоньес Ф.А., Кларамунт-Табернер Д., Хиль-Пенья Х. (декабрь 2015 г.). «Клинико-лабораторные подходы в диагностике ацидоза почечных канальцев». Детская нефрология. 30 (12): 2099–107. Дои:10.1007 / s00467-015-3083-9. PMID 25823989.
- ^ Сулеймани М., Растегар А. (сентябрь 2016 г.). «Патофизиология почечного тубулярного ацидоза: основной учебный план 2016». Американский журнал болезней почек. 68 (3): 488–98. Дои:10.1053 / j.ajkd.2016.03.422. PMID 27188519.
- ^ Pereira PC, Miranda DM, Oliveira EA, Silva AC (март 2009 г.). «Молекулярная патофизиология ацидоза почечных канальцев». Текущая геномика. 10 (1): 51–9. Дои:10.2174/138920209787581262. ЧВК 2699831. PMID 19721811.
- ^ Родригес Сориано Дж, Бойчис Х, Старк Х, Эдельманн С.М. (март 1967 г.). «Проксимальный почечный канальцевый ацидоз. Нарушение реабсорбции бикарбонатов при нормальном закислении мочи». Педиатрические исследования. 1 (2): 81–98. Дои:10.1203/00006450-196703000-00001. PMID 6029811.
- ^ Родригес Сориано Дж. (Август 2002 г.). «Почечный канальцевый ацидоз: клиническая картина». Журнал Американского общества нефрологов. 13 (8): 2160–70. Дои:10.1097 / 01.ASN.0000023430.92674.E5. PMID 12138150.
- ^ de Jong PE, Koomans HA, Weening JJ (2000). Klinische Nefrologie (3-е изд.). Маарссен: Эльзевир. С. 141–2.
- ^ Слай В.С., Уайт М.П., Сундарам В., Ташиан Р.Э., Хьюетт-Эммет Д., Гибо П. и др. (Июль 1985 г.). «Дефицит карбоангидразы II в 12 семьях с аутосомно-рецессивным синдромом остеопетроза с почечным канальцевым ацидозом и церебральной кальцификацией». Медицинский журнал Новой Англии. 313 (3): 139–45. Дои:10.1056 / NEJM198507183130302. PMID 3925334.
- ^ Шах Г. Н., Бонапас Г., Ху П. Я., Стришуглио П., Слай В. С. (сентябрь 2004 г.). «Синдром дефицита карбоангидразы II (остеопетроз с почечным канальцевым ацидозом и кальцификацией головного мозга): новые мутации в СА2, выявленные прямым секвенированием, расширяют возможности для корреляции генотип-фенотип». Человеческая мутация. 24 (3): 272. Дои:10.1002 / humu.9266. PMID 15300855.
- ^ Пушкин А., Абуладзе Н., Гросс Э., Ньюман Д., Татищев С., Ли И. и др. (Август 2004 г.). «Молекулярный механизм взаимодействия kNBC1-карбоангидраза II в клетках проксимальных канальцев». Журнал физиологии. 559 (Pt 1): 55–65. Дои:10.1113 / jphysiol.2004.065110. ЧВК 1665076. PMID 15218065.
- ^ Фатхаллах Д.М., Беджауи М., Лепаслье Д., Чейтер К., Слай В.С., Деллаги К. (май 1997 г.). «Дефицит карбоангидразы II (CA II) у пациентов из Магриба: доказательства эффекта основателя и геномной рекомбинации в локусе CA II». Генетика человека. 99 (5): 634–7. Дои:10.1007 / s004390050419. PMID 9150731.
- ^ а б Гипоренинемический гипоальдостеронизм в eMedicine
- ^ Лайтвуд Р. (1935). «Сообщение №1». Arch Dis Child. 10 (57): 205–6. Дои:10.1136 / adc.10.57.205. ЧВК 1975385.
- ^ Батлер AM, Уилсон JL, Фарбер S (1936). «Обезвоживание и ацидоз с обызвествлением почечных канальцев». Журнал педиатрии. 8 (4): 489–99. Дои:10.1016 / s0022-3476 (36) 80111-5.
- ^ Бейнс AM, Барелей Дж.А., Кук В.Т. (1945). «Нефрокальциноз, связанный с гиперхлоремией и низким содержанием бикарбоната в плазме». Q J Med. 14: 113–23.
- ^ Льюис DW (декабрь 1992 г.). «Что случилось с Крошечным Тимом?». Американский журнал болезней детей. 146 (12): 1403–7. Дои:10.1001 / архпеди.1992.02160240013002. PMID 1340779.
- ^ Альварес Дж., Себальос ФК, Квинтейро С. (15 апреля 2009 г.). «Роль инбридинга в исчезновении европейской королевской династии». PLOS One. 4 (4): e5174. Bibcode:2009PLoSO ... 4.5174A. Дои:10.1371 / journal.pone.0005174. ЧВК 2664480. PMID 19367331.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |