Аденинфосфорибозилтрансфераза - Adenine phosphoribosyltransferase




Аденинфосфорибозилтрансфераза (APRTase) является фермент закодировано APRT ген, нашел в люди на хромосома 16.[5] Он является частью семейства PRTase типа I и участвует в спасение нуклеотидов путь, который обеспечивает альтернативу нуклеотид биосинтез de novo у человека и большинства других животных.[6] В паразитарных простейшие такие как лямблии, APRTase обеспечивает единственный механизм, с помощью которого аденин могут быть произведены.[7] Дефицит APRTase способствует образованию камней в почках (мочекаменная болезнь ) и потенциальному почечная недостаточность.[8]

Функция
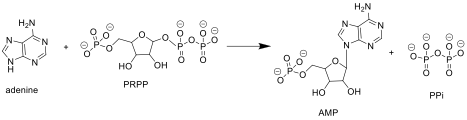
APRTase катализирует следующую реакцию в пурине спасение нуклеотидов путь:
Аденин + Фосфорибозилпирофосфат (PRPP ) → Аденилат (AMP ) + Пирофосфат (PPi )
В организмах, способных синтезировать пурины de novo, путь спасения нуклеотидов предоставляет альтернативу, которая является энергетически более эффективной. Он может спасти аденин от полиамин путь биосинтеза или из пищевых источников пуринов.[6] Хотя APRTase функционально избыточна в этих организмах, она становится более важной в периоды быстрого роста, такие как эмбриогенез и рост опухоли.[9] Он конститутивно экспрессируется во всех тканях млекопитающих.[10]
В простейшие паразитов, путь спасения нуклеотидов является единственным средством для синтеза нуклеотидов. Поскольку последствия дефицита APRTase у людей сравнительно легкие и поддаются лечению, можно лечить некоторые паразитарные инфекции путем нацеливания на функцию APRTase.[11]
В растения, как и у других организмов, ARPTase функционирует в первую очередь для синтеза аденилат. Обладает уникальной способностью метаболизировать цитокинины —А гормон растения что может существовать как основание, нуклеотид, или же нуклеозид —В аденилатных нуклеотидах.[12]
APRT функционально связан с гипоксантин-гуанинфосфорибозилтрансфераза (HPRT).
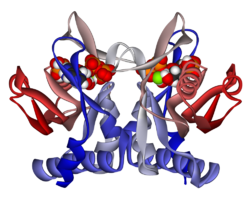
Структура
APRTase - это гомодимер, с 179 аминокислота остатки на мономер. Каждый мономер содержит следующие области:
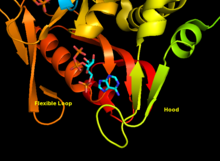
- «Core» домен (остатки 33-169) с пятью параллельными β-листы
- «Худ» домен (остатки 5-34) с 2 α-спирали и 2 β-листа
- Домен «гибкая петля» (остатки 95-113) с 2 антипараллельными β-листами[10]
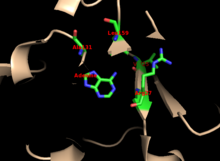
Ядро высоко консервативно во многих PRTases. Капюшон, в котором находится аденин сайт привязки, имеет большую вариабельность в семействе ферментов. Мотив из 13 остатков включает PRPP связывающая область и включает два соседних кислый остатки и по крайней мере одно окружающее гидрофобный остаток.[13]
Специфичность фермента к аденину включает гидрофобные остатки. Ala131 и Leu159 в основном домене. У человека два остатка в домене капота водородная связь с пурином для дальнейшей конкретности: Val25 с водород на N6, и Arg27 с N1. Хотя гибкая петля не взаимодействует с капюшоном во время распознавания пуринов, считается, что она закрывает активный сайт и изолировать реакцию от растворители.[10]
Большинство исследований APRTase сообщает, что Mg2+ необходим для переноса фосфорибозила, и он сохраняется в PRTases типа I.[12] Однако недавняя попытка определить структуру APRTase человека не смогла найти ни одного сайта для Mg2+, но нашли доказательства, позволяющие предположить, что Cl− атом около Trp98. Несмотря на сложность размещения Mg2+, принято считать, что каталитический механизм зависит от этого иона.[6]
Механизм
APRTase действует по двунаправленному последовательному механизму, включающему образование тройного комплекса. Фермент сначала связывает PRPP, с последующим аденин. После переноса фосфорибозила пирофосфат уходит первым, а затем AMP. Кинетические исследования показывают, что перенос фосфорибозила происходит относительно быстро, в то время как высвобождение продукта (особенно высвобождение АМФ) происходит довольно быстро. ограничение скорости.[9]
Считается, что в человеческой APRTase протон N9 аденина отводится Glu104 с образованием оксакарбения переходное состояние. Это функционирует как нуклеофил атаковать аномерный углерод PRPP, образующий AMP и вытесняющий пирофосфат из PRPP. Механизм APRTase в целом согласуется с механизмом других PRTases, которые сохраняют функцию замещения α-1-пирофосфата PRPP с использованием азот нуклеофил, либо в SN1 или SN2 атака.[6]
Дефицит
Когда активность APRTase снижена или отсутствует, аденин накапливается из других путей. Это ухудшается ксантиндегидрогеназа к 2,8-дигидроксиаденин (ДГК). Хотя DHA связывается с белками в плазма, у него плохой растворимость в моча и постепенно осаждается в почечные канальцы, приводящие к образованию камней в почках (мочекаменная болезнь ). Если не лечить, состояние может в конечном итоге вызвать почечная недостаточность.[8]
Дефицит ARPTase впервые был диагностирован в Великобритания в 1976 г. С тех пор у людей были определены две категории недостаточности APRTase.[14]
Дефицит типа I приводит к полной потере активности APRTase и может возникать у пациентов, которые гомозиготный или составной гетерозиготный для различных мутации.[15] Последовательность действий выявил множество различных мутаций, которые могут объяснить Тип 1, в том числе миссенс-мутации, бессмысленные мутации, дублированный набор из 4 пар оснований в экзон 3,[16] и один тимин вставка в интрон 4.[17] Эти мутации вызывают эффекты, которые сгруппированы в три основные области: связывание β-фосфата PRPP, связывание 5'-фосфата PRPP и сегмент гибкой петли, которая закрывается над активным центром во время катализа. [10] Дефицит I типа наблюдался у различных этнических групп, но изучался преимущественно среди белый населения.[17]
Дефицит типа II вызывает снижение сродства APRTase к PRPP, что приводит к десятикратному увеличению KM ценить.[6] Это наблюдалось и изучалось в основном в Япония.[17]
Диагноз дефицита APRTase может быть поставлен путем анализа камни в почках, измерение концентрации DHA в моче или анализ активности APRTase в эритроциты. Лечится регулярными дозами аллопуринол или фебуксостат, которые ингибируют активность ксантиндегидрогеназы, предотвращая накопление и осаждение DHA.[18] Состояние также можно смягчить с помощью диеты с низким содержанием пуринов и большого количества жидкости.[14]
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000198931 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск ансамбля 89: ENSMUSG00000006589 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ «Ссылка на Mouse PubMed:». Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Valaperta R, Rizzo V, Lombardi F, Verdelli C, Piccoli M, Ghiroldi A, Creo P, Colombo A, Valisi M, Margiotta E, Panella R, Costa E (1 июля 2014 г.). «Дефицит аденинфосфорибозилтрансферазы (APRT): идентификация новой бессмысленной мутации». BMC Нефрология. 15: 102. Дои:10.1186/1471-2369-15-102. ЧВК 4094445. PMID 24986359.
- ^ а б c d е Сильва Ч., Сильва М., Юлек Дж, Тиманн Огайо (июнь 2008 г.). «Структурные комплексы аденинфосфорибозилтрансферазы человека раскрывают новые особенности каталитического механизма APRT». Журнал биомолекулярной структуры и динамики. 25 (6): 589–97. Дои:10.1080/07391102.2008.10507205. PMID 18399692. S2CID 40788077.
- ^ Сарвер А.Е., Ван СС (октябрь 2002 г.). «Аденинфосфорибозилтрансфераза из Giardia lamblia имеет уникальный механизм реакции и необычные свойства связывания субстрата». Журнал биологической химии. 277 (42): 39973–80. Дои:10.1074 / jbc.M205595200. PMID 12171924.
- ^ а б Ши В., Танака К.С., Кротер Т.Р., Тейлор М.В., Алмо СК, Шрамм В.Л. (сентябрь 2001 г.). «Структурный анализ аденинфосфорибозилтрансферазы из Saccharomyces cerevisiae». Биохимия. 40 (36): 10800–9. Дои:10.1021 / bi010465h. PMID 11535055.
- ^ а б Башор К., Дену Дж. М., Бреннан Р. Г., Ульман Б. (март 2002 г.). «Кинетический механизм аденинфосфорибозилтрансферазы из Leishmania donovani». Биохимия. 41 (12): 4020–31. Дои:10.1021 / bi0158730. PMID 11900545.
- ^ а б c d Сильва М., Сильва С.Х., Юлек Дж., Тиманн Огайо (июнь 2004 г.). «Трехмерная структура аденинфосфорибозилтрансферазы человека и ее связь с DHA-мочекаменной болезнью». Биохимия. 43 (24): 7663–71. Дои:10.1021 / bi0360758. PMID 15196008.
- ^ Ши В., Сарвер А.Е., Ван СС, Танака К.С., Альмо СК, Шрамм В.Л. (октябрь 2002 г.). «Комплексы с закрытым сайтом аденинфосфорибозилтрансферазы из Giardia lamblia раскрывают механизм миграции рибозила». Журнал биологической химии. 277 (42): 39981–8. Дои:10.1074 / jbc.M205596200. PMID 12171925.
- ^ а б Аллен М., Цинь В., Моро Ф., Моффат Б. (май 2002 г.). «Изоформы аденинфосфорибозилтрансферазы Arabidopsis и их потенциальный вклад в метаболизм аденина и цитокининов». Physiologia Plantarum. 115 (1): 56–68. Дои:10.1034 / j.1399-3054.2002.1150106.x. PMID 12010467.
- ^ Лю Кью, Хироно С., Моригути I. (август 1990 г.). «Количественные отношения структура-активность для ингибиторов кальмодулина». Химико-фармацевтический бюллетень. 38 (8): 2184–9. Дои:10.1248 / cpb.38.2184. PMID 2279281.
- ^ а б Кэссиди MJ, McCulloch T, Fairbanks LD, Simmonds HA (март 2004 г.). «Диагностика дефицита аденинфосфорибозилтрансферазы как основной причины почечной недостаточности у реципиента почечного трансплантата». Нефрология, Диализ, Трансплантация. 19 (3): 736–8. Дои:10.1093 / ndt / gfg562. PMID 14767036.
- ^ Bollée G, Harambat J, Bensman A, Knebelmann B, Daudon M, Ceballos-Picot I (сентябрь 2012 г.). «Дефицит аденинфосфорибозилтрансферазы». Клинический журнал Американского общества нефрологов. 7 (9): 1521–7. Дои:10.2215 / CJN.02320312. PMID 22700886.
- ^ Каматани Н., Хакода М., Оцука С., Йошикава Х., Кашивадзаки С. (июль 1992 г.). «Только три мутации объясняют почти все дефектные аллели, вызывающие дефицит аденинфосфорибозилтрансферазы у японских пациентов». Журнал клинических исследований. 90 (1): 130–5. Дои:10.1172 / JCI115825. ЧВК 443071. PMID 1353080.
- ^ а б c Bollée G, Dollinger C, Boutaud L, Guillemot D, Bensman A, Harambat J, Deteix P, Daudon M, Knebelmann B, Ceballos-Picot I (апрель 2010 г.). «Фенотип и характеристика генотипа недостаточности аденинфосфорибозилтрансферазы». Журнал Американского общества нефрологов. 21 (4): 679–88. Дои:10.1681 / ASN.2009080808. ЧВК 2844298. PMID 20150536.
- ^ Эдвардссон В.О., Палссон Р., Сахота А. (1993). Пагон Р.А., Адам М.П., Ардингер Х.Х., Уоллес С.Е., Амемия А., Бин Л.Дж., Берд Т.Д., Фонг С.Т., Меффорд ХК, Смит Р.Дж., Стивенс К. (ред.). «Дефицит аденинфосфорибозилтрансферазы». SourceGeneReviews. PMID 22934314.
дальнейшее чтение
- Тишфилд Дж. А., Энгл С. Дж., Гупта П. К., Бай С., Бояджиев С., Шао С., О'Нил П., Альбертини Р. Дж., Стамбрук П. Дж., Сахота А. С. (1995). «Зародышевые и соматические мутации в локусе APRT мышей и человека». Достижения экспериментальной медицины и биологии. 370: 661–4. Дои:10.1007/978-1-4615-2584-4_137. ISBN 978-1-4613-6105-3. PMID 7660991.
- Такеучи Х., Канеко Й., Фудзита Дж., Йошида О. (апрель 1993 г.). «Случай сложной гетерозиготы при недостаточности аденинфосфорибозилтрансферазы (APRT * J / APRT * Q0), приводящей к 2,8-дигидроксиадениновой мочекаменной болезни: обзор зарегистрированных случаев с 2,8-дигидроксиадениновыми камнями в Японии». Журнал урологии. 149 (4): 824–6. Дои:10.1016 / s0022-5347 (17) 36222-5. PMID 8455250.
- Людвиг Х., Кузьмиц Р., Пичманн Х., Мюллер М.М. (ноябрь 1979 г.). «Ферменты системы взаимопревращения пуринов при хроническом лимфатическом лейкозе: снижение активности пуриновой нуклеозидфосфорилазы и аденозиндезаминазы». Blut. 39 (5): 309–15. Дои:10.1007 / BF01014193. PMID 116697. S2CID 6283377.
- Джонсон Л.А., Гордон Р.Б., Эммерсон Б.Т. (апрель 1977 г.). «Аденинфосфорибозилтрансфераза: простой спектрофотометрический анализ и частота мутаций в нормальной популяции». Биохимическая генетика. 15 (3–4): 265–72. Дои:10.1007 / BF00484458. PMID 869896. S2CID 41264715.
- Каматани Н., Хакода М., Оцука С., Йошикава Х., Кашивадзаки С. (июль 1992 г.). «Только три мутации объясняют почти все дефектные аллели, вызывающие дефицит аденинфосфорибозилтрансферазы у японских пациентов». Журнал клинических исследований. 90 (1): 130–5. Дои:10.1172 / JCI115825. ЧВК 443071. PMID 1353080.
- Чен Дж., Сахота А., Лаксдал Т., Скрин М., Боуман С., Цуй С., Стамбрук П. Дж., Тишфилд Дж. А. (декабрь 1991 г.). «Идентификация единственной миссенс-мутации в гене аденинфосфорибозилтрансферазы (APRT) у пяти исландских пациентов и одного британского пациента». Американский журнал генетики человека. 49 (6): 1306–11. ЧВК 1686459. PMID 1746557.
- Мимори А., Хидака Ю., Ву В.К., Тарле С.А., Каматани Н., Келли В.Н., Паллела Т.Д. (январь 1991 г.). «Мутантный аллель, характерный для дефицита аденинфосфорибозилтрансферазы I типа у японцев». Американский журнал генетики человека. 48 (1): 103–7. ЧВК 1682758. PMID 1985452.
- Чен Дж., Сахота А., Стамбрук П. Дж., Тишфилд Дж. А. (июль 1991 г.). «Амплификация полимеразной цепной реакции и анализ последовательности генов мутантной аденинфосфорибозилтрансферазы человека: природа и частота ошибок, вызванных ДНК-полимеразой Taq». Мутационные исследования. 249 (1): 169–76. Дои:10.1016 / 0027-5107 (91) 90143-С. PMID 2067530.
- Гатхоф Б.С., Сахота А., Грессер Ю., Чен Дж., Стамбрук П. Дж., Тишфилд Дж. А., Цёлльнер Н. (декабрь 1990 г.). «Идентификация мутации сплайсинга в локусе аденинфосфорибозилтрансферазы в немецкой семье». Klinische Wochenschrift. 69 (24): 1152–5. Дои:10.1007 / BF01815434. PMID 2135300. S2CID 11791868.
- Каматани Н., Куросима С., Хакода М., Палелла Т.Д., Хидака И. (октябрь 1990 г.). «Кроссоверы в короткой последовательности ДНК указывают на долгую эволюционную историю мутации APRT * J» (PDF). Генетика человека. 85 (6): 600–4. Дои:10.1007 / BF00193582. HDL:2027.42/47628. PMID 2227951. S2CID 10595601.
- Каматани Н., Куросима С., Тераи С., Хидака И., Палелла Т.Д., Нисиока К. (август 1989 г.). «Обнаружение аминокислотной замены в мутантном ферменте для особого типа дефицита аденинфосфорибозилтрансферазы (APRT) путем расщепления специфичного для последовательности белка». Американский журнал генетики человека. 45 (2): 325–31. ЧВК 1683345. PMID 2502918.
- Хидака Ю., Тарле С.А., Фухимори С., Каматани Н., Келли В.Н., Палелла Т.Д. (март 1988 г.). «Дефицит аденинфосфорибозилтрансферазы человека. Демонстрация единственного мутантного аллеля, общего для японцев». Журнал клинических исследований. 81 (3): 945–50. Дои:10.1172 / JCI113408. ЧВК 442550. PMID 3343350.
- Уилсон Дж. М., О'Тул Т. Э., Аргос П., Шуах Д. С., Даддона П. Е., Келли В. Н. (октябрь 1986 г.). «Аденинфосфорибозилтрансфераза человека. Полная аминокислотная последовательность фермента эритроцитов». Журнал биологической химии. 261 (29): 13677–83. PMID 3531209.
- Broderick TP, Schaff DA, Bertino AM, Dush MK, Tischfield JA, Stambrook PJ (май 1987 г.). «Сравнительная анатомия человеческого гена и фермента APRT: дивергенция нуклеотидной последовательности и сохранение неслучайного динуклеотидного расположения CpG». Труды Национальной академии наук Соединенных Штатов Америки. 84 (10): 3349–53. Дои:10.1073 / pnas.84.10.3349. ЧВК 304867. PMID 3554238.
- Хидака Ю., Палелла Т.Д., О'Тул Т.Э., Тарле С.А., Келли В.Н. (ноябрь 1987 г.). «Аденинфосфорибозилтрансфераза человека. Выявление аллельных мутаций на нуклеотидном уровне как причины полного дефицита фермента». Журнал клинических исследований. 80 (5): 1409–15. Дои:10.1172 / JCI113219. ЧВК 442397. PMID 3680503.
- Хидака Ю., Тарле С.А., О'Тул Т.Э., Келли В.Н., Палелла Т.Д. (ноябрь 1987 г.). «Нуклеотидная последовательность гена APRT человека». Исследования нуклеиновых кислот. 15 (21): 9086. Дои:10.1093 / nar / 15.21.9086. ЧВК 306432. PMID 3684585.
- Чен Дж., Сахота А., Мартин Г. Ф., Хакода М., Каматани Н., Стамбрук П. Дж., Тишфилд Дж. А. (июнь 1993 г.). «Анализ зародышевой линии и соматических мутаций in vivo в гене аденинфосфорибозилтрансферазы человека: горячие точки мутаций в донорском сайте сплайсинга интрона 4 и в кодоне 87». Мутационные исследования. 287 (2): 217–25. Дои:10.1016/0027-5107(93)90014-7. PMID 7685481.
- Сахота А., Чен Дж., Бояджиев С.А., Голт М.Х., Тишфилд Дж. А. (май 1994 г.). «Миссенс-мутация в гене аденинфосфорибозилтрансферазы, вызывающая уролитиаз 2,8-дигидроксиаденин». Молекулярная генетика человека. 3 (5): 817–8. Дои:10,1093 / чмг / 3,5,817. PMID 7915931.
внешняя ссылка
- Аденин + фосфорибозилтрансфераза в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)
- Человек APRT расположение генома и APRT страница сведений о генах в Браузер генома UCSC.
Галерея PDB | |
|---|---|
|