Рентгеновская кристаллография - X-ray crystallography

Рентгеновская кристаллография (XRC) является экспериментальной наукой, определяющей атомную и молекулярную структуру кристалл, в котором кристаллическая структура вызывает падающий луч Рентгеновские лучи к преломлять во многих конкретных направлениях. Измеряя углы и интенсивность этих дифрагированных лучей, кристаллограф может дать трехмерное изображение плотности электроны внутри кристалла. Из этого электронная плотность можно определить средние положения атомов в кристалле, а также их химические связи, их кристаллографический беспорядок, и другая различная информация.
Поскольку многие материалы могут образовывать кристаллы, например соли, металлы, минералы, полупроводники, а также различные неорганические, органические и биологические молекулы - рентгеновская кристаллография сыграла фундаментальную роль в развитии многих научных областей. В первые десятилетия использования этот метод определял размер атомов, длину и типы химических связей, а также различия в атомном масштабе между различными материалами, особенно минералами и минералами. сплавы. Метод также выявил структуру и функции многих биологических молекул, в том числе витамины, наркотики, белки и нуклеиновые кислоты Такие как ДНК. Рентгеновская кристаллография по-прежнему является основным методом описания атомной структуры новых материалов и выявления материалов, которые кажутся похожими на другие. эксперименты. рентгеновский снимок кристаллические структуры может также объяснить необычные электронный или же эластичный свойства материала, проливают свет на химические взаимодействия и процессы или служат основой для разработка фармацевтических препаратов против болезней.
При измерении дифракции рентгеновских лучей на монокристалле кристалл устанавливается на гониометр. Гониометр используется для позиционирования кристалла в выбранной ориентации. Кристалл подсвечивается тонко сфокусированным монохромный пучок рентгеновских лучей, производящий дифракционная картина регулярно расположенных пятен, известных как размышления. Двумерные изображения, полученные при разной ориентации, преобразуются в трехмерную модель плотности электронов внутри кристалла с использованием математического метода Преобразования Фурье в сочетании с химическими данными, известными для образца. Плохое разрешение (нечеткость) или даже ошибки могут возникнуть, если кристаллы слишком малы или недостаточно однородны по своему внутреннему составу.
Рентгеновская кристаллография связана с несколькими другими методами определения атомных структур. Подобные дифракционные картины могут быть получены путем рассеяния электронов или нейтроны, которые также интерпретируются Преобразование Фурье. Если монокристаллы достаточного размера не могут быть получены, могут быть применены различные другие рентгеновские методы для получения менее подробной информации; такие методы включают дифракция волокна, порошковая дифракция и (если образец не кристаллизовался) малоугловое рассеяние рентгеновских лучей (SAXS) .Если исследуемый материал доступен только в форме нанокристаллический порошков или страдает плохой кристалличностью, методы электронная кристаллография может применяться для определения атомной структуры.
Для всех вышеупомянутых методов дифракции рентгеновских лучей рассеяние эластичный; рассеянные рентгеновские лучи имеют то же длина волны как поступающий рентгеновский снимок. Напротив, неэластичный Методы рассеяния рентгеновских лучей полезны при изучении возбуждений образца, таких как плазмоны, кристаллическое поле и орбитальные возбуждения, магноны, и фононы, а не распределение его атомов.[1]
История
Ранняя научная история кристаллов и рентгеновских лучей

Кристаллы, хотя долгое время восхищались их регулярностью и симметрией, не исследовались научно до 17 века. Иоганн Кеплер предположил в своей работе Strena seu de Nive Sexangula (Новогодний подарок в виде шестиугольного снега) (1611 г.), что шестиугольная симметрия кристаллы снежинки произошло за счет регулярной упаковки сферических частиц воды.[2]

Датский ученый Николя Стено (1669) впервые провел экспериментальные исследования симметрии кристаллов. Стено показал, что углы между гранями одинаковы в каждом экземпляре кристалла определенного типа,[3] и Рене Жюст Хаю (1784) обнаружили, что каждую грань кристалла можно описать простым набором блоков одинаковой формы и размера. Следовательно, Уильям Хэллоуз Миллер в 1839 году смог присвоить каждой грани уникальную метку из трех маленьких целых чисел, Индексы Миллера которые до сих пор используются для идентификации граней кристаллов. Исследование Хайя привело к правильной идее, что кристаллы представляют собой регулярный трехмерный массив ( Решетка Браве ) атомов и молекулы; один ячейка повторяется бесконечно по трем основным направлениям, которые не обязательно перпендикулярны. В XIX веке полный каталог возможных симметрий кристалла был разработан Йохан Хессель,[4] Огюст Браве,[5] Евграф Федоров,[6] Артур Шёнфлис[7] и (с опозданием) Уильям Барлоу (1894 г.). На основе имеющихся данных и физических соображений Барлоу предложил несколько кристаллических структур в 1880-х годах, которые позже были подтверждены рентгеновской кристаллографией;[8] однако доступных данных в 1880-х годах было слишком мало, чтобы считать его модели убедительными.
Вильгельм Рентген открыл рентгеновские лучи в 1895 году, когда исследования симметрии кристаллов были завершены. Физики не были уверены в природе рентгеновских лучей, но вскоре заподозрили, что это волны электромагнитное излучение, форма свет. В Максвелл теория электромагнитное излучение был хорошо принят среди ученых, и эксперименты Чарльз Гловер Баркла показали, что рентгеновские лучи демонстрируют явления, связанные с электромагнитными волнами, в том числе поперечные поляризация и спектральные линии сродни наблюдаемым в видимом диапазоне длин волн. Однощелевые эксперименты в лаборатории Арнольд Зоммерфельд предположил, что рентгеновские лучи длина волны около 1 ангстрем. Рентгеновские лучи - это не только волны, но и фотоны, и обладают свойствами частиц. Альберт Эйнштейн представил концепцию фотона в 1905 году,[9] но это не было широко распространено до 1922 г.,[10][11] когда Артур Комптон подтвердил это рассеянием рентгеновских лучей на электронах.[12] Подобные частицам свойства рентгеновских лучей, такие как ионизация газов, побудили Уильям Генри Брэгг в 1907 году утверждать, что рентгеновские лучи нет электромагнитное излучение.[13][14][15][16] Точка зрения Брэгга оказалась непопулярной, и наблюдение дифракция рентгеновских лучей к Макс фон Лауэ в 1912 г.[17] подтвердил для большинства ученых, что рентгеновские лучи являются формой электромагнитного излучения.
дифракция рентгеновских лучей
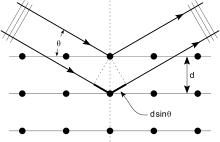
Кристаллы представляют собой регулярные массивы атомов, а рентгеновские лучи можно рассматривать как волны электромагнитного излучения. Атомы рассеивают рентгеновские волны, в основном, через электроны атомов. Подобно тому, как океанская волна, ударяющаяся о маяк, производит вторичные круговые волны, исходящие от маяка, так и рентгеновские лучи, падающие на электрон, создают вторичные сферические волны, исходящие от него. Это явление известно как упругое рассеяние, а электрон (или маяк) известен как рассеиватель. Регулярный массив рассеивателей создает регулярный массив сферических волн. Хотя эти волны нейтрализуют друг друга в большинстве направлений через деструктивное вмешательство, они конструктивно складываются в нескольких конкретных направлениях, определенных Закон Брэгга:

Здесь d - расстояние между дифрагирующими плоскостями, - угол падения, п - любое целое число, а λ - длина волны луча. Эти конкретные направления отображаются в виде точек на дифракционная картина называется размышления. Таким образом, дифракция рентгеновских лучей возникает в результате попадания электромагнитной волны (рентгеновских лучей) на регулярный массив рассеивателей (повторяющееся расположение атомов внутри кристалла).

Рентгеновские лучи используются для создания дифракционной картины, потому что их длина волны λ обычно того же порядка величины (1–100 ангстрем), что и расстояние d между плоскостями в кристалле. В принципе, любая волна, падающая на регулярный массив рассеивателей, производит дифракция, как сначала предсказал Франческо Мария Гримальди в 1665 г. Для получения значительной дифракции расстояние между рассеивателями и длина волны падающей волны должны быть одинаковыми по размеру. Для иллюстрации, дифракция солнечного света через птичье перо впервые была описана Джеймс Грегори в конце 17 века. Первый искусственный дифракционные решетки для видимого света были построены Дэвид Риттенхаус в 1787 г. и Йозеф фон Фраунгофер в 1821 году. Однако видимый свет имеет слишком большую длину волны (обычно 5500 ангстрем), чтобы наблюдать дифракцию на кристаллах. До первых экспериментов по дифракции рентгеновских лучей расстояния между плоскостями решетки в кристалле не были известны с уверенностью.
Идея использования кристаллов в качестве дифракционная решетка за Рентгеновские лучи возникла в 1912 году в разговоре между Пол Питер Эвальд и Макс фон Лауэ в Английский сад в Мюнхен. Эвальд предложил модель кристаллов резонатора для своей диссертации, но эту модель нельзя было проверить с помощью видимый свет, так как длина волны была намного больше, чем расстояние между резонаторами. Фон Лауэ понял, что для наблюдения таких малых расстояний необходимо электромагнитное излучение с более короткой длиной волны, и предположил, что рентгеновские лучи могут иметь длину волны, сравнимую с расстоянием между элементарными ячейками в кристаллах. Фон Лауэ работал с двумя техниками, Вальтером Фридрихом и его помощником Полем Книппингом, чтобы направить луч рентгеновского излучения через сульфат меди кристалл и запишите его дифракцию на фотопластинка. После проявления пластина показала большое количество четко определенных пятен, расположенных в виде пересекающихся кругов вокруг пятна, созданного центральным лучом.[17][18] Фон Лауэ разработал закон, который связывает углы рассеяния, размер и ориентацию расстояний между элементарными ячейками в кристалле, за что он был награжден премией Нобелевская премия по физике в 1914 г.[19]
Рассеяние
Как описано в математический вывод ниже, рассеяние рентгеновских лучей определяется плотностью электронов внутри кристалла. Поскольку энергия рентгеновского излучения намного больше, чем энергия валентного электрона, рассеяние можно смоделировать как Томсоновское рассеяние, взаимодействие электромагнитного луча со свободным электроном. Эта модель обычно применяется для описания поляризации рассеянного излучения.
Интенсивность Томсоновское рассеяние для одной частицы с массой м и элементарный заряд q является:[20]

Следовательно, атомные ядра, которые намного тяжелее электрона, вносят незначительный вклад в рассеянное рентгеновское излучение.
Развитие с 1912 по 1920 год

После новаторских исследований фон Лауэ эта область быстро развивалась, в первую очередь физиками. Уильям Лоуренс Брэгг и его отец Уильям Генри Брэгг. В 1912–1913 годах Брэгг-младший разработал Закон Брэгга, который связывает наблюдаемое рассеяние с отражениями от равномерно расположенных плоскостей внутри кристалла.[21][22][23] Брэгги, отец и сын, разделили Нобелевскую премию 1915 года по физике за свои работы в области кристаллографии. Самые ранние конструкции в целом были простыми и отличались одномерной симметрией. Однако по мере совершенствования вычислительных и экспериментальных методов в течение следующих десятилетий стало возможным вывести надежные атомные позиции для более сложных двух- и трехмерных расположений атомов в элементарной ячейке.
Потенциал рентгеновской кристаллографии для определения структуры молекул и минералов - тогда еще смутно известный из химических и гидродинамических экспериментов - был реализован сразу. Самые ранние структуры были простыми неорганическими кристаллами и минералами, но даже они раскрывали фундаментальные законы физики и химии. Первой структурой с атомным разрешением, которая была «решена» (то есть определена) в 1914 году, была структура столовая соль.[24][25][26] Распределение электронов в структуре поваренной соли показало, что кристаллы не обязательно состоят из ковалентно связанный молекул, и доказал существование ионные соединения.[27] Структура алмаз было решено в том же году,[28][29] доказывая тетраэдрическое расположение его химических связей и показывая, что длина одинарной связи C – C составляла 1,52 ангстрем. Другие ранние структуры включены медь,[30] фторид кальция (CaF2, также известный как флюорит), кальцит (CaCO3) и пирит (FeS2)[31] в 1914 г .; шпинель (MgAl2О4) в 1915 г .;[32][33] то рутил и анатаз формы оксид титана (TiO2) в 1916 г .;[34] пирохроит Mn (OH)2 и, соответственно, брусит Mg (OH)2 в 1919 г.[35][36] Также в 1919 г. нитрат натрия (NaNO3) и дихлоройодид цезия (CsICl2) были определены Ральф Уолтер Грейстоун Вайкофф, а вюрцит Структура (гексагональный ZnS) стала известна в 1920 году.[37]
Структура графит была решена в 1916 г.[38] родственным методом порошковая дифракция,[39] который был разработан Питер Дебай и Пол Шеррер и, независимо, Альберт Халл в 1917 г.[40] Структура графита была определена методом дифракции на монокристаллах в 1924 г. двумя группами независимо друг от друга.[41][42] Халл также использовал порошковый метод для определения структуры различных металлов, таких как железо.[43] и магний.[44]
Культурное и эстетическое значение
В 1951 году группа Фестивального выкройки на Фестиваль Британии организовал совместную группу производителей текстиля и опытных кристаллографов для разработки кружев и принтов на основе рентгеновской кристаллографии инсулин, фарфоровая глина, и гемоглобин. Одним из ведущих ученых проекта был Др. Хелен Мегоу (1907–2002), в то время помощник директора по исследованиям в Кавендишской лаборатории в Кембридже. Мегоу считается одной из центральных фигур, которая черпала вдохновение из кристаллических диаграмм и увидела их потенциал в дизайне.[45] В 2008 году Wellcome Collection в Лондоне организовала выставку Festival Pattern Group под названием «От атома к узорам».[45]
Вклад в химию и материаловедение
Рентгеновская кристаллография позволила лучше понять химические связи и нековалентные взаимодействия. Первоначальные исследования выявили типичные радиусы атомов и подтвердили многие теоретические модели химической связи, такие как тетраэдрическая связь углерода в структуре алмаза,[28] октаэдрическая связь металлов, наблюдаемая в гексахлороплатинате (IV) аммония,[46] и резонанс, наблюдаемый в планарной карбонатной группе[31] и в ароматических молекулах.[47] Кэтлин Лонсдейл структура 1928 г. гексаметилбензол[48] установили гексагональную симметрию бензол и показал четкое различие в длине связи между алифатическими связями C – C и ароматическими связями C – C; это открытие привело к идее резонанс между химическими связями, которые имели глубокие последствия для развития химии.[49] Ее выводы были предвосхищены Уильям Генри Брэгг, опубликовавшие модели нафталин и антрацен в 1921 году на основе других молекул, ранняя форма молекулярная замена.[47][50]
Также в 1920-х гг. Виктор Мориц Гольдшмидт и позже Линус Полинг разработаны правила исключения химически маловероятных структур и определения относительных размеров атомов. Эти правила привели к структуре Brookite (1928) и понимание относительной стабильности рутил, Brookite и анатаз формы оксид титана.
Расстояние между двумя связанными атомами является чувствительной мерой прочности связи и ее ордер на облигации; таким образом, рентгеновские кристаллографические исследования привели к открытию еще более экзотических типов связи в неорганическая химия, такие как двойные связи металл-металл,[51][52][53] металл-металл четверные связи,[54][55][56] и трехцентровые, двухэлектронные связи.[57] Рентгеновская кристаллография - или, строго говоря, неупругая Комптоновское рассеяние эксперимент - также предоставил доказательства частично ковалентного характера водородные связи.[58] В области металлоорганическая химия, рентгеновская структура ферроцен инициировал научные исследования сэндвич-смеси,[59][60] в то время как Соль Цейзе стимулировали исследования в области "обратного связывания" и комплексов металл-пи.[61][62][63][64] Наконец, рентгеновская кристаллография сыграла новаторскую роль в развитии супрамолекулярная химия, особенно в выяснении структуры краун-эфиры и принципы химия между хозяином и гостем.
Рентгеновская дифракция - очень мощный инструмент в катализатор разработка. Измерения ex-situ обычно проводятся для проверки кристаллической структуры материалов или для обнаружения новых структур. In-situ эксперименты дают полное представление о структурной стабильности катализаторов в условиях реакции.[65][66][67]
В материаловедении много сложных неорганический и металлоорганический системы были проанализированы с использованием монокристаллических методов, таких как фуллерены, металлопорфирины, и другие сложные соединения. Монокристаллическая дифракция также используется в фармацевтическая индустрия, из-за недавних проблем с полиморфы. Основными факторами, влияющими на качество монокристаллических структур, являются размер кристалла и его форма; перекристаллизация является широко используемым методом для улучшения этих факторов в кристаллах с небольшими молекулами. В Кембриджская структурная база данных содержит более 1000000 структур по состоянию на июнь 2019 года; более 99% этих структур были определены методом дифракции рентгеновских лучей.
Минералогия и металлургия

С 1920-х годов дифракция рентгеновских лучей была основным методом определения расположения атомов в минералах и минералах. металлы. Применение рентгеновской кристаллографии в минералогия началось со структуры гранат, который был определен в 1924 году Менцером. Систематическое рентгеноструктурное исследование силикаты была предпринята в 1920-х годах. Это исследование показало, что, поскольку Si /О Когда соотношение изменяется, кристаллы силиката обнаруживают значительные изменения в их атомном расположении. Мачачки распространил эти идеи на минералы, в которых алюминий заменители кремний атомы силикатов. Первое применение рентгеновской кристаллографии в металлургия То же произошло и в середине 1920-х годов.[69][70][71][72][73][74] В частности, Линус Полинг структура сплава Mg2Sn[75] привел к его теории устойчивости и структуры сложных ионных кристаллов.[76]
17 октября 2012 г. Марсоход Curiosity на планета марс в "Rocknest »провели первый рентгеноструктурный анализ Марсианский грунт. Результаты марсохода CheMin анализатор выявили наличие нескольких минералов, в том числе полевой шпат, пироксены и оливин, и предположил, что марсианский грунт в образце был похож на «выветрившийся» базальтовые почвы " из Гавайские вулканы.[68]
Ранние органические и малые биологические молекулы
Первая структура органического соединения, гексаметилентетрамин, была решена в 1923 году.[77] За этим последовало несколько исследований длинноцепочечных жирные кислоты, которые являются важным компонентом биологические мембраны.[78][79][80][81][82][83][84][85][86] В 1930-х годах начали выясняться структуры гораздо более крупных молекул с двумерной сложностью. Значительным достижением стала структура фталоцианин,[87] большая плоская молекула, которая тесно связана с молекулы порфирина важно в биологии, например гем, Коррин и хлорофилл.
Рентгеновская кристаллография биологических молекул взлетела с Дороти Кроуфут Ходжкин, решивших структуры холестерин (1937), пенициллин (1946) и витамин B12 (1956), за что была награждена Нобелевская премия по химии в 1964 г. В 1969 г. ей удалось решить структуру инсулин, над которым она работала более тридцати лет.[88]
Биологическая кристаллография макромолекул

Кристаллические структуры белков (которые нерегулярны и в сотни раз больше, чем холестерин) начали решаться в конце 1950-х годов, начиная со структуры кашалот миоглобин к Сэр Джон Каудери Кендрю,[89] за что он поделился Нобелевская премия по химии с Макс Перуц в 1962 г.[90] После этого успеха было определено более 130 000 рентгеновских кристаллических структур белков, нуклеиновых кислот и других биологических молекул.[91] Ближайшим конкурирующим методом по количеству проанализированных структур является спектроскопия ядерного магнитного резонанса (ЯМР), который разрешил менее одной десятой от многих.[92] Кристаллография может определять структуры произвольно больших молекул, тогда как ЯМР в состоянии раствора ограничивается относительно маленькими (менее 70 kДа ). Рентгеновская кристаллография обычно используется для определения того, как фармацевтический препарат взаимодействует со своей белковой мишенью и какие изменения могут его улучшить.[93] Однако внутренняя мембранные белки по-прежнему сложно кристаллизоваться, потому что для них требуются моющие средства или другие денатурирующие средства чтобы солюбилизировать их изолированно, и такие детергенты часто препятствуют кристаллизации. Мембранные белки являются важным компонентом геном, и включают в себя многие белки, имеющие большое физиологическое значение, такие как ионные каналы и рецепторы.[94][95] Криогеника гелия используются для предотвращения радиационного повреждения кристаллов белка.[96]
С другой стороны, даже относительно небольшие молекулы могут создавать проблемы для разрешающей способности рентгеновской кристаллографии. Структура, присвоенная в 1991 г. антибиотику, выделенному из морского организма, диазонамид А (C40ЧАС34Cl2N6О6, молярная масса 765,65 г / моль), оказалась неверной классическим доказательством структуры: синтетический образец не был идентичен натуральному продукту. Ошибка была приписана неспособности рентгеновской кристаллографии различить правильные -OH / -NH и замененные -NH2 / -O- группы в неправильной структуре.[97] Однако с развитием приборостроения аналогичные группы можно выделить с помощью современных монокристаллических рентгеновских дифрактометров.
Несмотря на то, что это бесценный инструмент в структурная биология, кристаллография белков несет в себе некоторые проблемы в своей методологии, которые затрудняют интерпретацию данных. Кристаллическая решетка, которая образуется в процессе кристаллизации, содержит множество единиц очищенного белка, которые плотно и симметрично упакованы в кристалле. При поиске ранее неизвестного белка выяснение его формы и границ внутри кристаллической решетки может быть сложной задачей. Белки обычно состоят из более мелких субъединиц, и задача различения субъединиц и идентификации реального белка может быть сложной задачей даже для опытных кристаллографов. Небиологические интерфейсы, которые возникают во время кристаллизации, известны как контакты упаковки кристаллов (или просто контакты кристаллов) и не могут быть определены кристаллографическими средствами. Когда новая структура белка решена с помощью рентгеновской кристаллографии и депонирована в Банк данных белков, его авторов просят указать «биологическую сборку», которая будет составлять функциональный, биологически значимый белок. Однако ошибки, недостающие данные и неточные аннотации во время представления данных приводят к неясным структурам и ставят под угрозу надежность базы данных. Сообщается, что уровень ошибок только в случае ошибочных аннотаций превышает 6,6%.[98] или примерно 15%,[99] возможно, нетривиальный размер, учитывая количество депонированных структур. Эта «проблема классификации интерфейсов» обычно решается с помощью вычислительных подходов и стала признанной темой в структурная биоинформатика.
Методы рассеивания
Упругое и неупругое рассеяние
Рентгеновская кристаллография - это форма упругое рассеяние; исходящие рентгеновские лучи имеют ту же энергию и, следовательно, ту же длину волны, что и входящие рентгеновские лучи, только с измененным направлением. Напротив, неупругое рассеяние происходит, когда энергия передается от входящего рентгеновского излучения к кристаллу, например, путем возбуждения электрона внутренней оболочки к более высокому уровень энергии. Такое неупругое рассеяние снижает энергию (или увеличивает длину волны) выходящего луча. Неупругое рассеяние полезно для исследования таких возбуждений вещества, но не для определения распределения рассеивателей в веществе, что является целью рентгеновской кристаллографии.
Рентгеновские лучи диапазон длин волн от 10 до 0,01 нанометры; типичная длина волны, используемая для кристаллографии, равна 1Å (0,1 нм),[100] что находится на шкале ковалентных химические связи и радиус одиночного атома. Более длинноволновые фотоны (например, ультрафиолетовый радиация ) не будет иметь достаточного разрешения для определения положения атомов. С другой стороны, фотоны с более короткой длиной волны, такие как гамма излучение их трудно производить в больших количествах, их трудно сфокусировать, и они слишком сильно взаимодействуют с материей, производя пары частица-античастица. Следовательно, рентгеновские лучи являются «сладким пятном» для длины волны при определении структур с атомным разрешением по рассеянию электромагнитное излучение.
Другие рентгенологические методы
Другие формы упругого рассеяния рентгеновских лучей, помимо дифракции на монокристаллах, включают: порошковая дифракция, Малоугловое рассеяние рентгеновских лучей (SAXS ) и несколько видов рентгеновского дифракция волокна, который использовался Розалинд Франклин в определении двойная спираль из ДНК. В общем, дифракция рентгеновских лучей на монокристаллах дает больше структурной информации, чем эти другие методы; однако для этого требуется достаточно большой и обычный кристалл, который не всегда доступен.
Эти методы рассеяния обычно используют монохромный Рентгеновские лучи, которые ограничены одной длиной волны с небольшими отклонениями. Широкий спектр рентгеновских лучей (то есть смесь рентгеновских лучей с разными длинами волн) также может использоваться для проведения дифракции рентгеновских лучей, метод, известный как метод Лауэ. Это метод, использованный в первоначальном открытии дифракции рентгеновских лучей. Рассеяние Лауэ дает много структурной информации только при коротком воздействии рентгеновского луча, и поэтому используется в структурных исследованиях очень быстрых событий (Кристаллография с временным разрешением ). Однако он не так хорошо подходит, как монохроматическое рассеяние, для определения полной атомной структуры кристалла и поэтому лучше работает с кристаллами с относительно простым расположением атомов.
Режим обратного отражения Лауэ регистрирует рентгеновские лучи, рассеянные назад от источника широкого спектра. Это полезно, если образец слишком толстый для прохождения через него рентгеновских лучей. Плоскости дифрагирования в кристалле определяются, зная, что нормаль к плоскости дифрагирования делит угол между падающим лучом и дифрагированным лучом пополам. А Диаграмма Гренингера может быть использован[101] интерпретировать фотографию заднего отражения Лауэ.
Электронная и нейтронная дифракция
Другие частицы, такие как электроны и нейтроны, может использоваться для создания дифракционная картина. Хотя рассеяние электронов, нейтронов и рентгеновских лучей основано на разных физических процессах, полученные дифракционные картины анализируются с использованием тех же когерентная дифракционная визуализация техники.
Как показано ниже, электронная плотность внутри кристалла и дифрактограммы связаны простым математическим методом: преобразование Фурье, что позволяет относительно легко рассчитывать плотность по рисункам. Однако это работает, только если рассеяние слабый, т.е. если рассеянные пучки намного менее интенсивны, чем приходящий пучок. Слабо рассеянные пучки проходят через остальную часть кристалла, не подвергаясь второму рассеянию. Такие повторно рассеянные волны называются «вторичным рассеянием» и затрудняют анализ. Любой достаточно толстый кристалл будет вызывать вторичное рассеяние, но поскольку рентгеновские лучи относительно слабо взаимодействуют с электронами, это обычно не вызывает серьезного беспокойства. Напротив, электронные лучи могут вызывать сильное вторичное рассеяние даже для относительно тонких кристаллов (> 100 нм). Так как эта толщина соответствует диаметру многих вирусы перспективным направлением является электронография изолированных макромолекулярные сборки, Такие как популярный капсиды и молекулярные машины, который может быть проведен с помощью криогенногоэлектронный микроскоп. Кроме того, сильное взаимодействие электронов с веществом (примерно в 1000 раз сильнее, чем для рентгеновских лучей) позволяет определять атомную структуру чрезвычайно малых объемов. Область применения для электронная кристаллография варьируется от биомолекул, таких как мембранные белки, на тонких органических пленках до сложных структур (нанокристаллических) интерметаллических соединений и цеолитов.
Дифракция нейтронов - отличный метод для определения структуры, хотя было трудно получить интенсивные монохроматические пучки нейтронов в достаточных количествах. Традиционно ядерные реакторы были использованы, хотя источники, производящие нейтроны раскол становятся все более доступными. Будучи незаряженными, нейтроны гораздо легче рассеиваются на ядрах атомов, чем на электронах. Следовательно, рассеяние нейтронов очень полезно для наблюдения положения легких атомов с небольшим количеством электронов, особенно водород, который практически не виден при дифракции рентгеновских лучей. Рассеяние нейтронов также имеет замечательное свойство, заключающееся в том, что растворитель можно сделать невидимым, регулируя соотношение нормальных воды, H2O и тяжелая вода, D2О.
Методы
Обзор дифракции рентгеновских лучей на монокристаллах

Самый старый и точный метод рентгена кристаллография является дифракция рентгеновских лучей на монокристаллах, в котором пучок рентгеновских лучей попадает на монокристалл, создавая рассеянные пучки. Когда они попадают на кусок пленки или другой детектор, эти лучи дифракционная картина пятен; сила и углы этих лучей записываются по мере постепенного поворота кристалла.[102] Каждое место называется отражение, поскольку он соответствует отражению рентгеновских лучей от одного набора равномерно расположенных плоскостей внутри кристалла. Для монокристаллов достаточной чистоты и регулярности данные дифракции рентгеновских лучей могут определять среднюю длину и углы химической связи с точностью до нескольких тысячных долей ангстрема и до нескольких десятых долей ангстрема. степень, соответственно. Атомы в кристалле не статичны, а колеблются вокруг своего среднего положения, обычно менее чем на несколько десятых ангстрема. Рентгеновская кристаллография позволяет измерить величину этих колебаний.
Процедура
Методика рентгеновской кристаллографии монокристаллов состоит из трех основных этапов. Первый - и часто самый трудный - шаг - получить адекватный кристалл исследуемого материала. Кристалл должен быть достаточно большим (обычно более 0,1 мм по всем размерам), чистым по составу и правильной по структуре, без значительных внутренних несовершенства такие как трещины или побратимство.
На втором этапе кристалл помещается в интенсивный пучок рентгеновских лучей, обычно с одной длиной волны (монохроматические рентгеновские лучи), создавая правильную картину отражений. Измеряются углы и интенсивности дифрагированных рентгеновских лучей, при этом каждое соединение имеет уникальную дифракционную картину.[103] По мере того, как кристалл постепенно поворачивается, предыдущие отражения исчезают и появляются новые; интенсивность каждого пятна записывается при каждой ориентации кристалла. Может потребоваться сбор нескольких наборов данных, каждый из которых охватывает чуть более половины полного оборота кристалла и обычно содержит десятки тысяч отражений.
На третьем этапе эти данные вычислительно комбинируются с дополнительной химической информацией для создания и уточнения модели расположения атомов внутри кристалла. Окончательная, усовершенствованная модель атомного устройства, теперь называемая Кристальная структура - обычно хранится в общедоступной базе данных.
Ограничения
По мере того, как повторяющаяся единица кристалла, его элементарная ячейка, становится больше и сложнее, картина атомного уровня, обеспечиваемая рентгеновской кристаллографией, становится менее разрешенной (более "нечеткой") для данного числа наблюдаемых отражений. Часто различают два предельных случая рентгеновской кристаллографии - «низкомолекулярная» (которая включает сплошные неорганические твердые тела) и «макромолекулярная» кристаллография. Кристаллография малых молекул обычно включает кристаллы, содержащие менее 100 атомов в их асимметричный блок; такие кристаллические структуры обычно настолько хорошо разрешены, что атомы можно различить как изолированные «капли» электронной плотности. Напротив, макромолекулярная кристаллография часто включает в себя десятки тысяч атомов в элементарной ячейке. Такие кристаллические структуры обычно менее разрешены (более «размыты»); атомы и химические связи выглядят как трубки электронной плотности, а не как изолированные атомы. Как правило, небольшие молекулы также легче кристаллизовать, чем макромолекулы; однако рентгеновская кристаллография оказалась возможной даже для вирусы и белки с сотнями тысяч атомов, благодаря улучшенной кристаллографической визуализации и технологии.[104] Хотя обычно рентгеновская кристаллография может выполняться только в том случае, если образец находится в кристаллической форме, были проведены новые исследования по отбору образцов некристаллических форм.[105]
Кристаллизация

Хотя кристаллографию можно использовать для характеристики беспорядка в нечистом или неправильном кристалле, для кристаллографии обычно требуется чистый кристалл высокой регулярности, чтобы определить структуру сложного расположения атомов. Pure, regular crystals can sometimes be obtained from natural or synthetic materials, such as samples of металлы, minerals or other macroscopic materials. The regularity of such crystals can sometimes be improved with macromolecular crystal отжиг[106][107][108] и другие методы. However, in many cases, obtaining a diffraction-quality crystal is the chief barrier to solving its atomic-resolution structure.[109]
Small-molecule and macromolecular crystallography differ in the range of possible techniques used to produce diffraction-quality crystals. Small molecules generally have few degrees of conformational freedom, and may be crystallized by a wide range of methods, such as химическое осаждение из паровой фазы и перекристаллизация. By contrast, macromolecules generally have many degrees of freedom and their crystallization must be carried out so as to maintain a stable structure. For example, proteins and larger РНК molecules cannot be crystallized if their tertiary structure has been unfolded; therefore, the range of crystallization conditions is restricted to solution conditions in which such molecules remain folded.
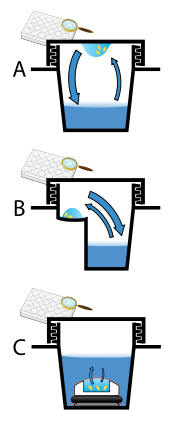
Protein crystals are almost always grown in solution. The most common approach is to lower the solubility of its component molecules very gradually; if this is done too quickly, the molecules will precipitate from solution, forming a useless dust or amorphous gel on the bottom of the container. Crystal growth in solution is characterized by two steps: nucleation of a microscopic crystallite (possibly having only 100 molecules), followed by рост of that crystallite, ideally to a diffraction-quality crystal.[110][111] The solution conditions that favor the first step (nucleation) are not always the same conditions that favor the second step (subsequent growth). The crystallographer's goal is to identify solution conditions that favor the development of a single, large crystal, since larger crystals offer improved resolution of the molecule. Consequently, the solution conditions should disfavor the first step (nucleation) but favor the second (growth), so that only one large crystal forms per droplet. If nucleation is favored too much, a shower of small crystallites will form in the droplet, rather than one large crystal; if favored too little, no crystal will form whatsoever. Other approaches involves, crystallizing proteins under oil, where aqueous protein solutions are dispensed under liquid oil, and water evaporates through the layer of oil. Different oils have different evaporation permeabilities, therefore yielding changes in concentration rates from different percipient/protein mixture.[112]
It is extremely difficult to predict good conditions for nucleation or growth of well-ordered crystals.[113] In practice, favorable conditions are identified by скрининг; a very large batch of the molecules is prepared, and a wide variety of crystallization solutions are tested.[114] Hundreds, even thousands, of solution conditions are generally tried before finding the successful one. The various conditions can use one or more physical mechanisms to lower the solubility of the molecule; for example, some may change the pH, some contain salts of the Серия Хофмайстера or chemicals that lower the dielectric constant of the solution, and still others contain large polymers such as полиэтиленгликоль that drive the molecule out of solution by entropic effects. It is also common to try several temperatures for encouraging crystallization, or to gradually lower the temperature so that the solution becomes supersaturated. These methods require large amounts of the target molecule, as they use high concentration of the molecule(s) to be crystallized. Due to the difficulty in obtaining such large quantities (milligrams ) of crystallization-grade protein, robots have been developed that are capable of accurately dispensing crystallization trial drops that are in the order of 100 nanoliters in volume. This means that 10-fold less protein is used per experiment when compared to crystallization trials set up by hand (in the order of 1 микролитр ).[115]
Several factors are known to inhibit or mar crystallization. The growing crystals are generally held at a constant temperature and protected from shocks or vibrations that might disturb their crystallization. Impurities in the molecules or in the crystallization solutions are often inimical to crystallization. Conformational flexibility in the molecule also tends to make crystallization less likely, due to entropy. Molecules that tend to self-assemble into regular helices are often unwilling to assemble into crystals.[нужна цитата ] Crystals can be marred by побратимство, which can occur when a unit cell can pack equally favorably in multiple orientations; although recent advances in computational methods may allow solving the structure of some twinned crystals. Having failed to crystallize a target molecule, a crystallographer may try again with a slightly modified version of the molecule; even small changes in molecular properties can lead to large differences in crystallization behavior.
Сбор информации
Mounting the crystal

The crystal is mounted for measurements so that it may be held in the X-ray beam and rotated. There are several methods of mounting. In the past, crystals were loaded into glass capillaries with the crystallization solution (the маточный раствор ). Nowadays, crystals of small molecules are typically attached with oil or glue to a glass fiber or a loop, which is made of nylon or plastic and attached to a solid rod. Protein crystals are scooped up by a loop, then flash-frozen with жидкий азот.[116] This freezing reduces the radiation damage of the X-rays, as well as the noise in the Bragg peaks due to thermal motion (the Debye-Waller effect). However, untreated protein crystals often crack if flash-frozen; therefore, they are generally pre-soaked in a cryoprotectant solution before freezing.[117] Unfortunately, this pre-soak may itself cause the crystal to crack, ruining it for crystallography. Generally, successful cryo-conditions are identified by trial and error.
The capillary or loop is mounted on a goniometer, which allows it to be positioned accurately within the X-ray beam and rotated. Since both the crystal and the beam are often very small, the crystal must be centered within the beam to within ~25 micrometers accuracy, which is aided by a camera focused on the crystal. The most common type of goniometer is the "kappa goniometer", which offers three angles of rotation: the ω angle, which rotates about an axis perpendicular to the beam; the κ angle, about an axis at ~50° to the ω axis; and, finally, the φ angle about the loop/capillary axis. When the κ angle is zero, the ω and φ axes are aligned. The κ rotation allows for convenient mounting of the crystal, since the arm in which the crystal is mounted may be swung out towards the crystallographer. The oscillations carried out during data collection (mentioned below) involve the ω axis only. An older type of goniometer is the four-circle goniometer, and its relatives such as the six-circle goniometer.
X-ray sources
Rotating anode
Small scale crystallography can be done with a local Рентгеновская трубка source, typically coupled with an image plate детектор. These have the advantage of being relatively inexpensive and easy to maintain, and allow for quick screening and collection of samples. However, the wavelength of the light produced is limited by the availability of different анод материалы. Furthermore, the intensity is limited by the power applied and cooling capacity available to avoid melting the anode. In such systems, electrons are boiled off of a cathode and accelerated through a strong electric potential of ~50кВ; having reached a high speed, the electrons collide with a metal plate, emitting тормозное излучение and some strong spectral lines corresponding to the excitation of inner-shell electrons металла. The most common metal used is медь, which can be kept cool easily, due to its high теплопроводность, and which produces strong Kα и Kβ линий. The Kβ line is sometimes suppressed with a thin (~10 µm) nickel foil. The simplest and cheapest variety of sealed X-ray tube has a stationary anode (the Crookes tube ) and run with ~2 кВт of electron beam power. The more expensive variety has a rotating-anode type source that run with ~14 kW of e-beam power.
X-rays are generally filtered (by use of X-ray filters ) to a single wavelength (made monochromatic) and коллимированный to a single direction before they are allowed to strike the crystal. The filtering not only simplifies the data analysis, but also removes radiation that degrades the crystal without contributing useful information. Collimation is done either with a collimator (basically, a long tube) or with a clever arrangement of gently curved mirrors. Mirror systems are preferred for small crystals (under 0.3 mm) or with large unit cells (over 150 Å).
Rotating anodes were used by Joanna (Joka) Maria Vandenberg in the first experiments[118][119] that demonstrated the power of X rays for quick (in real time production) screening of large InGaAsP тонкая пленка wafers for контроль качества из quantum well lasers.
Синхротронное излучение
Синхротронное излучение sources are some of the brightest light sources on earth and are some of the most powerful tools available to X-ray crystallographers. X-ray beams generated in large machines called синхротроны which accelerate electrically charged particles, often electrons, to nearly the speed of light and confine them in a (roughly) circular loop using magnetic fields.
Synchrotrons are generally national facilities, each with several dedicated beamlines where data is collected without interruption. Synchrotrons were originally designed for use by high-energy physicists studying субатомные частицы и космический явления. The largest component of each synchrotron is its electron storage ring. This ring is actually not a perfect circle, but a many-sided polygon. At each corner of the polygon, or sector, precisely aligned magnets bend the electron stream. As the electrons' path is bent, they emit bursts of energy in the form of X-rays.
Using synchrotron radiation frequently has specific requirements for X-ray crystallography. Интенсивный ионизирующего излучения can cause радиационное повреждение to samples, particularly macromolecular crystals. Cryo crystallography protects the sample from radiation damage, by freezing the crystal at жидкий азот temperatures (~100 K ).[120] However, synchrotron radiation frequently has the advantage of user-selectable wavelengths, allowing for anomalous scattering experiments which maximizes anomalous signal. This is critical in experiments such as ГРУСТНЫЙ и СУМАСШЕДШИЙ.
Лазер на свободных электронах
Лазеры на свободных электронах have been developed for use in X-ray crystallography.[121] These are the brightest X-ray sources currently available; with the X-rays coming in фемтосекунда bursts. The intensity of the source is such that atomic resolution diffraction patterns can be resolved for crystals otherwise too small for collection. However, the intense light source also destroys the sample,[122] requiring multiple crystals to be shot. As each crystal is randomly oriented in the beam, hundreds of thousands of individual diffraction images must be collected in order to get a complete data set. This method, serial femtosecond crystallography, has been used in solving the structure of a number of protein crystal structures, sometimes noting differences with equivalent structures collected from synchrotron sources.[123]
Recording the reflections

When a crystal is mounted and exposed to an intense beam of X-rays, it scatters the X-rays into a pattern of spots or размышления that can be observed on a screen behind the crystal. A similar pattern may be seen by shining a лазерный указатель в компакт-диск. The relative intensities of these spots provide the information to determine the arrangement of molecules within the crystal in atomic detail. The intensities of these reflections may be recorded with фотопленка, an area detector (such as a pixel detector ) or with a устройство с зарядовой связью (CCD) image sensor. The peaks at small angles correspond to low-resolution data, whereas those at high angles represent high-resolution data; thus, an upper limit on the eventual resolution of the structure can be determined from the first few images. Some measures of diffraction quality can be determined at this point, such as the mosaicity of the crystal and its overall disorder, as observed in the peak widths. Some pathologies of the crystal that would render it unfit for solving the structure can also be diagnosed quickly at this point.
One image of spots is insufficient to reconstruct the whole crystal; it represents only a small slice of the full Fourier transform. To collect all the necessary information, the crystal must be rotated step-by-step through 180°, with an image recorded at every step; actually, slightly more than 180° is required to cover reciprocal space, due to the curvature of the Сфера Эвальда. However, if the crystal has a higher symmetry, a smaller angular range such as 90° or 45° may be recorded. The rotation axis should be changed at least once, to avoid developing a "blind spot" in reciprocal space close to the rotation axis. It is customary to rock the crystal slightly (by 0.5–2°) to catch a broader region of reciprocal space.
Multiple data sets may be necessary for certain phasing methods. For example, MAD phasing requires that the scattering be recorded at least three (and usually four, for redundancy) wavelengths of the incoming X-ray radiation. A single crystal may degrade too much during the collection of one data set, owing to radiation damage; in such cases, data sets on multiple crystals must be taken.[124]
Анализ данных
Crystal symmetry, unit cell, and image scaling
The recorded series of two-dimensional diffraction patterns, each corresponding to a different crystal orientation, is converted into a three-dimensional model of the electron density; the conversion uses the mathematical technique of Fourier transforms, which is explained ниже. Each spot corresponds to a different type of variation in the electron density; the crystallographer must determine который variation corresponds to который spot (индексация), the relative strengths of the spots in different images (merging and scaling) and how the variations should be combined to yield the total electron density (фазировка).
Data processing begins with индексация the reflections. This means identifying the dimensions of the unit cell and which image peak corresponds to which position in reciprocal space. A byproduct of indexing is to determine the symmetry of the crystal, i.e., its космическая группа. Some space groups can be eliminated from the beginning. For example, reflection symmetries cannot be observed in chiral molecules; thus, only 65 space groups of 230 possible are allowed for protein molecules which are almost always chiral. Indexing is generally accomplished using an autoindexing рутина.[125] Having assigned symmetry, the data is then интегрированный. This converts the hundreds of images containing the thousands of reflections into a single file, consisting of (at the very least) records of the Miller index of each reflection, and an intensity for each reflection (at this state the file often also includes error estimates and measures of partiality (what part of a given reflection was recorded on that image)).
A full data set may consist of hundreds of separate images taken at different orientations of the crystal. The first step is to merge and scale these various images, that is, to identify which peaks appear in two or more images (слияние) and to scale the relative images so that they have a consistent intensity scale. Optimizing the intensity scale is critical because the relative intensity of the peaks is the key information from which the structure is determined. The repetitive technique of crystallographic data collection and the often high symmetry of crystalline materials cause the diffractometer to record many symmetry-equivalent reflections multiple times. This allows calculating the symmetry-related R-factor, a reliability index based upon how similar are the measured intensities of symmetry-equivalent reflections,[требуется разъяснение ] thus assessing the quality of the data.
Initial phasing
The data collected from a diffraction experiment is a взаимное пространство representation of the crystal lattice. The position of each diffraction 'spot' is governed by the size and shape of the unit cell, and the inherent симметрия внутри кристалла. The intensity of each diffraction 'spot' is recorded, and this intensity is proportional to the square of the structure factor амплитуда. В structure factor это комплексное число containing information relating to both the амплитуда и фаза из волна. In order to obtain an interpretable electron density map, both amplitude and phase must be known (an electron density map allows a crystallographer to build a starting model of the molecule). The phase cannot be directly recorded during a diffraction experiment: this is known as the фазовая проблема. Initial phase estimates can be obtained in a variety of ways:
- Ab initio фазировка или же прямые методы – This is usually the method of choice for small molecules (<1000 non-hydrogen atoms), and has been used successfully to solve the phase problems for small proteins. If the resolution of the data is better than 1.4 Å (140 вечера ), прямые методы can be used to obtain phase information, by exploiting known phase relationships between certain groups of reflections.[126][127]
- Molecular replacement – if a related structure is known, it can be used as a search model in molecular replacement to determine the orientation and position of the molecules within the unit cell. The phases obtained this way can be used to generate electron density maps.[128]
- Аномальное рассеяние рентгеновских лучей (СУМАСШЕДШИЙ или же SAD phasing ) – the X-ray wavelength may be scanned past an absorption edge[когда определяется как? ] of an atom, which changes the scattering in a known way. By recording full sets of reflections at three different wavelengths (far below, far above and in the middle of the absorption edge) one can solve for the substructure of the anomalously diffracting atoms and hence the structure of the whole molecule. The most popular method of incorporating anomalous scattering atoms into proteins is to express the protein in a метионин auxotroph (a host incapable of synthesizing methionine) in a media rich in seleno-methionine, which contains селен атомы. A MAD experiment can then be conducted around the absorption edge, which should then yield the position of any methionine residues within the protein, providing initial phases.[129]
- Heavy atom methods (множественная изоморфная замена ) – If electron-dense metal atoms can be introduced into the crystal, прямые методы или же Patterson-space methods can be used to determine their location and to obtain initial phases. Such heavy atoms can be introduced either by soaking the crystal in a heavy atom-containing solution, or by co-crystallization (growing the crystals in the presence of a heavy atom). As in MAD phasing, the changes in the scattering amplitudes can be interpreted to yield the phases. Although this is the original method by which protein crystal structures were solved, it has largely been superseded by MAD phasing with selenomethionine.[128]
Model building and phase refinement
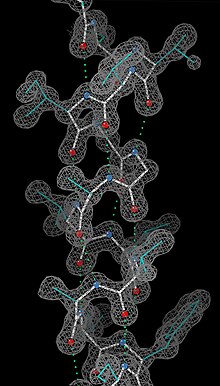

Having obtained initial phases, an initial model can be built. The atomic positions in the model and their respective Debye-Waller factors (или же B-factors, accounting for the thermal motion of the atom) can be refined to fit the observed diffraction data, ideally yielding a better set of phases. A new model can then be fit to the new electron density map and successive rounds of refinement is carried out. This interative process continues until the correlation between the diffraction data and the model is maximized. The agreement is measured by an р-фактор определяется как

куда F это structure factor. A similar quality criterion is рсвободный, which is calculated from a subset (~10%) of reflections that were not included in the structure refinement. Обе р factors depend on the resolution of the data. As a rule of thumb, рсвободный should be approximately the resolution in angstroms divided by 10; thus, a data-set with 2 Å resolution should yield a final рсвободный ~ 0.2. Chemical bonding features such as stereochemistry, hydrogen bonding and distribution of bond lengths and angles are complementary measures of the model quality. Phase bias is a serious problem in such iterative model building. Omit maps are a common technique used to check for this.[требуется разъяснение ]
It may not be possible to observe every atom in the asymmetric unit. In many cases, Кристаллографический беспорядок smears the electron density map. Weakly scattering atoms such as hydrogen are routinely invisible. It is also possible for a single atom to appear multiple times in an electron density map, e.g., if a protein sidechain has multiple (<4) allowed conformations. In still other cases, the crystallographer may detect that the covalent structure deduced for the molecule was incorrect, or changed. For example, proteins may be cleaved or undergo post-translational modifications that were not detected prior to the crystallization.
Беспорядок
A common challenge in refinement of crystal structures results from crystallographic disorder. Disorder can take many forms but in general involves the coexistence of two or more species or conformations. Failure to recognize disorder results in flawed interpretation. Pitfalls from improper modeling of disorder are illustrated by the discounted hypothesis of bond stretch isomerism.[132] Disorder is modelled with respect to the relative population of the components, often only two, and their identity. In structures of large molecules and ions, solvent and counterions are often disordered.
Applied computational data analysis
The use of computational methods for the powder X-ray diffraction data analysis is now generalized. It typically compares the experimental data to the simulated diffractogram of a model structure, taking into account the instrumental parameters, and refines the structural or microstructural parameters of the model using наименьших квадратов based minimization algorithm. Most available tools allowing phase identification and structural refinement are based on the Rietveld method,[133][134] some of them being open and free software such as FullProf Suite,[135][136] Jana2006,[137] MAUD,[138][139][140] Rietan,[141] GSAS,[142] etc. while others are available under commercials licenses such as Diffrac.Suite TOPAS,[143] Match!,[144] etc. Most of these tools also allow Le Bail refinement (also referred to as profile matching), that is, refinement of the cell parameters based on the Bragg peaks positions and peak profiles, without taking into account the crystallographic structure by itself. More recent tools allow the refinement of both structural and microstructural data, such as the FAULTS program included in the FullProf Suite,[145] which allows the refinement of structures with planar defects (e.g. stacking faults, twinnings, intergrowths).
Deposition of the structure
Once the model of a molecule's structure has been finalized, it is often deposited in a crystallographic database такой как Cambridge Structural Database (for small molecules), the Inorganic Crystal Structure Database (ICSD) (for inorganic compounds) or the Банк данных белков (for protein and sometimes nucleic acids). Many structures obtained in private commercial ventures to crystallize medicinally relevant proteins are not deposited in public crystallographic databases.
Diffraction theory
The main goal of X-ray crystallography is to determine the density of electrons ж(р) throughout the crystal, where р represents the three-dimensional position вектор внутри кристалла. To do this, X-ray scattering is used to collect data about its Fourier transform F(q), which is inverted mathematically to obtain the density defined in real space, using the formula

где интеграл is taken over all values of q. The three-dimensional real vector q represents a point in взаимное пространство, that is, to a particular oscillation in the electron density as one moves in the direction in which q точки. Длина q соответствует divided by the wavelength of the oscillation. The corresponding formula for a Fourier transform will be used below


где интеграл is summed over all possible values of the position vector р внутри кристалла.
The Fourier transform F(q) is generally a комплексное число, and therefore has a величина |F(q) | и фаза φ(q) related by the equation

The intensities of the reflections observed in X-ray diffraction give us the magnitudes |F(q) | but not the phases φ(q). To obtain the phases, full sets of reflections are collected with known alterations to the scattering, either by modulating the wavelength past a certain absorption edge or by adding strongly scattering (i.e., electron-dense) metal atoms such as Меркурий. Combining the magnitudes and phases yields the full Fourier transform F(q), which may be inverted to obtain the electron density ж(р).
Crystals are often idealized as being прекрасно периодический. In that ideal case, the atoms are positioned on a perfect lattice, the electron density is perfectly periodic, and the Fourier transform F(q) is zero except when q принадлежит к обратная решетка (так называемой Bragg peaks). In reality, however, crystals are not perfectly periodic; atoms vibrate about their mean position, and there may be disorder of various types, such as mosaicity, dislocations, разные точечные дефекты, and heterogeneity in the conformation of crystallized molecules. Therefore, the Bragg peaks have a finite width and there may be significant diffuse scattering, a continuum of scattered X-rays that fall between the Bragg peaks.
Intuitive understanding by Bragg's law
An intuitive understanding of X-ray diffraction can be obtained from the Bragg model of diffraction. In this model, a given reflection is associated with a set of evenly spaced sheets running through the crystal, usually passing through the centers of the atoms of the crystal lattice. The orientation of a particular set of sheets is identified by its three Miller indices (час, k, л), and let their spacing be noted by d. William Lawrence Bragg proposed a model in which the incoming X-rays are scattered specularly (mirror-like) from each plane; from that assumption, X-rays scattered from adjacent planes will combine constructively (конструктивное вмешательство ) when the angle θ between the plane and the X-ray results in a path-length difference that is an integer multiple п of the X-ray wavelength λ.

A reflection is said to be indexed when its Miller indices (or, more correctly, its обратная решетка vector components) have been identified from the known wavelength and the scattering angle 2θ. Such indexing gives the unit-cell parameters, the lengths and angles of the unit-cell, as well as its космическая группа. С Bragg's law does not interpret the relative intensities of the reflections, however, it is generally inadequate to solve for the arrangement of atoms within the unit-cell; for that, a Fourier transform method must be carried out.
Scattering as a Fourier transform
The incoming X-ray beam has a polarization and should be represented as a vector wave; however, for simplicity, let it be represented here as a scalar wave. We also ignore the complication of the time dependence of the wave and just concentrate on the wave's spatial dependence. Плоские волны can be represented by a волновой вектор kв, and so the strength of the incoming wave at time т = 0 is given by

At position р within the sample, let there be a density of scatterers ж(р); these scatterers should produce a scattered spherical wave of amplitude proportional to the local amplitude of the incoming wave times the number of scatterers in a small volume dV о р

куда S is the proportionality constant.
Consider the fraction of scattered waves that leave with an outgoing wave-vector of kиз and strike the screen at рэкран. Since no energy is lost (elastic, not inelastic scattering), the wavelengths are the same as are the magnitudes of the wave-vectors |kв|=|kиз|, From the time that the photon is scattered at р until it is absorbed at рэкран, the photon undergoes a change in phase

The net radiation arriving at рэкран is the sum of all the scattered waves throughout the crystal

which may be written as a Fourier transform

куда q = kиз – kв. The measured intensity of the reflection will be square of this amplitude

Friedel and Bijvoet mates
For every reflection corresponding to a point q in the reciprocal space, there is another reflection of the same интенсивность at the opposite point -q. This opposite reflection is known as the Friedel mate of the original reflection. This symmetry results from the mathematical fact that the density of electrons ж(р) at a position р всегда настоящий номер. Как указано выше, ж(р) is the inverse transform of its Fourier transform F(q); however, such an inverse transform is a комплексное число в целом. To ensure that ж(р) is real, the Fourier transform F(q) must be such that the Friedel mates F(−q) и F(q) находятся complex conjugates друг друга. Таким образом, F(−q) has the same magnitude as F(q) but they have the opposite phase, i.e., φ(q) = −φ(q)

The equality of their magnitudes ensures that the Friedel mates have the same intensity |F|2. Эта симметрия позволяет измерить полное преобразование Фурье только из половины обратного пространства, например, повернув кристалл чуть более чем на 180 ° вместо полного вращения на 360 °. В кристаллах со значительной симметрией даже большее количество отражений может иметь одинаковую интенсивность (Bijvoet mates); в таких случаях, возможно, потребуется измерить даже меньшую часть обратного пространства. В благоприятных случаях высокой симметрии иногда требуется только 90 ° или даже 45 ° данных, чтобы полностью исследовать обратное пространство.
Ограничение Фриделя-мате может быть получено из определения обратного преобразования Фурье

С Формула Эйлера заявляет, что еяИкс = cos (Икс) + я грешу (Икс) обратное преобразование Фурье можно разделить на сумму чисто действительной части и чисто мнимой части

Функция ж(р) действительна тогда и только тогда, когда второй интеграл ягрех равен нулю для всех значений р. В свою очередь, это верно тогда и только тогда, когда выполняется указанное выше ограничение.

поскольку ягрех = −ягрех подразумевает, что ягрех = 0.
Сфера Эвальда
Каждое изображение дифракции рентгеновских лучей представляет собой только срез, сферический срез обратного пространства, что можно увидеть по конструкции сферы Эвальда. Обе kиз и kв имеют одинаковую длину из-за упругого рассеяния, так как длина волны не изменилась. Следовательно, их можно представить как два радиальных вектора в сфере в взаимное пространство, где показаны значения q которые отбираются на данном дифракционном изображении. Поскольку имеется небольшой разброс в длинах волн входящего рентгеновского луча, значения |F(q) | можно измерить только при q векторы, расположенные между двумя сферами, соответствующими этим радиусам. Следовательно, чтобы получить полный набор данных преобразования Фурье, необходимо повернуть кристалл немного больше, чем на 180 °, а иногда и меньше, если присутствует достаточная симметрия. Полное вращение на 360 ° не требуется из-за симметрии, присущей преобразованиям Фурье реальных функций (таких как плотность электронов), но требуется «чуть больше» 180 °, чтобы покрыть все обратное пространство с заданным разрешением из-за кривизна Сфера Эвальда. На практике кристалл раскачивается на небольшую величину (0,25–1 °), чтобы учесть отражения у границ сферических оболочек Эвальда.
Функция Паттерсона
Хорошо известным результатом преобразований Фурье является автокорреляция теорема, которая утверждает, что автокорреляция c(р) функции ж(р)

имеет преобразование Фурье C(q), что является квадратом величины F(q)

Следовательно, автокорреляционная функция c(р) электронной плотности (также известной как Функция Паттерсона[146]) можно вычислить непосредственно из интенсивностей отражений, без вычисления фаз. В принципе, это можно использовать для непосредственного определения кристаллической структуры; однако на практике это сложно реализовать. Автокорреляционная функция соответствует распределению векторов между атомами в кристалле; таким образом, кристалл N атомы в своей элементарной ячейке могут иметь N(N - 1) пики функции Паттерсона. Учитывая неизбежные ошибки в измерении интенсивностей и математические трудности восстановления положений атомов по межатомным векторам, этот метод редко используется для решения структур, за исключением простейших кристаллов.
Преимущества кристалла
В принципе, атомную структуру можно определить, применяя рассеяние рентгеновских лучей к некристаллическим образцам, даже к отдельной молекуле. Однако кристаллы дают гораздо более сильный сигнал из-за своей периодичности. Кристаллический образец по определению является периодическим; кристалл состоит из многих элементарные ячейки повторяется бесконечно в трех независимых направлениях. Такие периодические системы имеют преобразование Фурье который сосредоточен в периодически повторяющихся точках обратного пространства, известного как Пики Брэгга; пики Брэгга соответствуют пятнам отражения, наблюдаемым на дифракционном изображении. Поскольку амплитуда этих отражений линейно растет с числом N рассеивателей наблюдаемые интенсивность этих пятен должны расти квадратично, как N2. Другими словами, использование кристалла концентрирует слабое рассеяние отдельных элементарных ячеек в гораздо более мощное когерентное отражение, которое можно наблюдать над шумом. Это пример конструктивное вмешательство.
В жидком, порошковом или аморфном образце молекулы внутри этого образца имеют случайную ориентацию. Такие образцы имеют непрерывный спектр Фурье, который равномерно расширяет его амплитуду, тем самым уменьшая интенсивность измеренного сигнала, как это наблюдается в SAXS. Что еще более важно, ориентировочная информация теряется. Хотя теоретически это возможно, экспериментально сложно получить структуры сложных асимметричных молекул с атомным разрешением из таких усредненных по вращению данных. Промежуточный случай дифракция волокна в котором субъединицы расположены периодически, по меньшей мере, в одном измерении.
Нобелевские премии по рентгеновской кристаллографии
| Год | Лауреат | Приз | Обоснование |
|---|---|---|---|
| 1914 | Макс фон Лауэ | Физика | «За открытие дифракции рентгеновских лучей на кристаллах»,[147] важный шаг в развитии Рентгеновская спектроскопия. |
| 1915 | Уильям Генри Брэгг | Физика | "За их услуги в анализе Кристальная структура с помощью рентгеновских лучей »[148] |
| 1915 | Уильям Лоуренс Брэгг | Физика | "За их услуги в анализе Кристальная структура с помощью рентгеновских лучей »[148] |
| 1962 | Макс Ф. Перуц | Химия | "за исследования структур глобулярные белки "[149] |
| 1962 | Джон К. Кендрю | Химия | "за исследования структур глобулярные белки "[149] |
| 1962 | Джеймс Дьюи Уотсон | Лекарство | "За открытия, касающиеся молекулярной структуры нуклеиновые кислоты и его значение для передачи информации в живом материале "[150] |
| 1962 | Фрэнсис Гарри Комптон Крик | Лекарство | "За открытия, касающиеся молекулярной структуры нуклеиновые кислоты и его значение для передачи информации в живом материале "[150] |
| 1962 | Морис Хью Фредерик Уилкинс | Лекарство | "За открытия, касающиеся молекулярной структуры нуклеиновые кислоты и его значение для передачи информации в живом материале "[150] |
| 1964 | Дороти Ходжкин | Химия | "Для нее определения рентгеновскими методами структур важных биохимических веществ »[151] |
| 1972 | Стэнфорд Мур | Химия | "За их вклад в понимание связи между химической структурой и каталитической активностью активного центра рибонуклеаза молекула "[152] |
| 1972 | Уильям Х. Штайн | Химия | "За их вклад в понимание связи между химической структурой и каталитической активностью активного центра рибонуклеаза молекула "[152] |
| 1976 | Уильям Н. Липскомб | Химия | "За исследования структуры бораны освещая проблемы химической связи »[153] |
| 1985 | Джером Карл | Химия | "За выдающиеся достижения в развитии прямые методы для определения кристаллических структур »[154] |
| 1985 | Герберт А. Хауптман | Химия | "За выдающиеся достижения в развитии прямые методы для определения кристаллических структур »[154] |
| 1988 | Иоганн Дайзенхофер | Химия | «За определение трехмерной структуры фотосинтетический реакционный центр "[155] |
| 1988 | Хартмут Мишель | Химия | «За определение трехмерной структуры фотосинтетический реакционный центр "[155] |
| 1988 | Роберт Хубер | Химия | «За определение трехмерной структуры фотосинтетический реакционный центр "[155] |
| 1997 | Джон Э. Уокер | Химия | "За разъяснение ферментативный механизм лежащий в основе синтеза аденозинтрифосфата (АТФ) »[156] |
| 2003 | Родерик Маккиннон | Химия | "За открытия, касающиеся каналов в клеточных мембранах [...] для структурных и механических исследования ионных каналов "[157] |
| 2003 | Питер Агре | Химия | "За открытие каналов в клеточных мембранах [...] за открытие водные каналы "[157] |
| 2006 | Роджер Д. Корнберг | Химия | "За исследования молекулярных основ эукариотическая транскрипция "[158] |
| 2009 | Ада Э. Йонат | Химия | «Для изучения структуры и функции рибосома "[159] |
| 2009 | Томас А. Стейтц | Химия | «Для изучения структуры и функции рибосома "[159] |
| 2009 | Венкатраман Рамакришнан | Химия | «Для изучения структуры и функции рибосома "[159] |
| 2012 | Брайан Кобылка | Химия | "Для изучения Рецепторы, связанные с G-белком "[160] |
Приложения
Рентгеновская дифракция имеет широкое и разнообразное применение в химических, биохимических, физических, материаловедческих и минералогических науках. Лауэ заявил в 1937 году, что эта техника «расширила возможности наблюдения мельчайших структур в десять тысяч раз по сравнению с тем, что дает нам микроскоп».[161] Рентгеновская дифракция аналогична микроскопу с разрешением на атомном уровне, который показывает атомы и их электронное распределение.
Рентгеновская дифракция, дифракция электронов и дифракция нейтронов дают информацию о структуре вещества, кристаллического и некристаллического, на атомном и молекулярном уровне. Кроме того, эти методы могут применяться при изучении свойств всех материалов, неорганических, органических или биологических. Из-за важности и разнообразия приложений дифракционных исследований кристаллов за такие исследования было присуждено множество Нобелевских премий.[162]
Идентификация лекарства
Рентгеновская дифракция была использована для идентификации антибиотиков, таких как: восемь β-лактам (ампициллин натрия, пенициллин G прокаин, цефалексин, тригидрат ампициллина, бензатин пенициллин, бензилпенициллин натрия, цефотаксим натрия, Цефтриаксон натрия ), три тетрациклин (доксициклина гидрохлорид, окситетрациклина дегидрат, тетрациклина гидрохлорид ) и два макролид (азитромицин, эстолат эритромицина ) антибиотические препараты. Каждый из этих препаратов имеет уникальный образец рентгеновской дифракции (XRD), который делает возможной их идентификацию.[163]
Характеристика текстильных волокон и полимеров
Судебно-медицинская экспертиза любых следовых доказательств основано на Принцип обмена Локара. Это гласит, что «каждый контакт оставляет след». На практике, даже если передача материала произошла, его может быть невозможно обнаружить, поскольку переданная сумма очень мала.[164]
Текстильные волокна представляют собой смесь кристаллических и аморфных веществ. Следовательно, измерение степени кристалличности дает полезные данные для характеристики волокон с помощью рентгеновской дифрактометрии. Сообщалось, что дифракция рентгеновских лучей использовалась для определения «кристаллического» осадка, обнаруженного на стуле. Отложение оказалось аморфным, но имеющаяся дифракционная картина соответствовала картине полиметилметакрилата. Пиролиз масс-спектрометрии позже идентифицировали месторождение как полиметилцианоакрилон параметров кристалла Боина.[165]
Исследование костей
Нагревание или сжигание костей вызывает заметные изменения в минерале кости, которые можно обнаружить с помощью рентгеноструктурных методов. В течение первых 15 минут нагревания при 500 ° C или выше костные кристаллы начали изменяться. При более высоких температурах толщина и форма кристаллов костей кажутся стабилизированными, но когда образцы нагревали при более низкой температуре или в течение более короткого периода, рентгеновские дифракционные рентгеновские лучи показали резкие изменения параметров кристаллов.[166]
Интегральные схемы
Рентгеновская дифракция была продемонстрирована как метод исследования сложной структуры интегральные схемы.[167]
Смотрите также
- Полоса Биверса – Липсона
- Брэгговская дифракция
- Кристаллографическая база данных
- Кристаллографические точечные группы
- Карта разницы плотности
- Электронная дифракция
- Энергодисперсионная дифракция рентгеновских лучей
- Параметр Flack
- Предел Хендерсона
- Международный год кристаллографии
- Джон Десмонд Бернал
- Формализм мультипольной плотности
- Нейтронная дифракция
- Порошковая дифракция
- Птихография
- Уравнение Шеррера
- Малоугловое рассеяние рентгеновских лучей (МУРР)
- Определение структуры
- Сверхбыстрый рентген
- Широкоугольное рентгеновское рассеяние (ВОСК)
Рекомендации
- ^ "Резонансное рассеяние рентгеновских лучей | Лаборатория Шена". arpes.stanford.edu. Получено 2019-07-10.
- ^ Кеплер Дж. (1611). Strena seu de Nive Sexangula. Франкфурт: Г. Тампах. ISBN 3-321-00021-0.
- ^ Steno N (1669). De solido intra solidum naturaliter contento is prodromus. Florentiae.
- ^ Гессель JFC (1831 г.). Kristallometrie oder Kristallonomie und Kristallographie. Лейпциг.
- ^ Браве А (1850). "Mémoire sur les systèmes formés par des points distribués regièrement sur un plan ou dans l'espace". Journal de l'École Polytechnique. 19: 1.
- ^ Шафрановский И.И., Белов Н.В. (1962). Пол Эвальд (ред.). "Э. С. Федоров" (PDF). 50 лет рентгеновской дифракции. Спрингер: 351. ISBN 90-277-9029-9.
- ^ Schönflies A (1891). Kristallsysteme und Kristallstruktur. Лейпциг.
- ^ Барлоу В. (1883 г.). «Вероятный характер внутренней симметрии кристаллов». Природа. 29 (738): 186. Bibcode:1883Натура..29..186Б. Дои:10.1038 / 029186a0. Смотрите также Барлоу, Уильям (1883). «Вероятная природа внутренней симметрии кристаллов». Природа. 29 (739): 205. Bibcode:1883Натура..29..205Б. Дои:10.1038 / 029205a0. Зонке, Л. (1884). «Вероятная природа внутренней симметрии кристаллов». Природа. 29 (747): 383. Bibcode:1884Натура..29..383С. Дои:10.1038 / 029383a0. S2CID 4072817. Барлоу, ВМ. (1884). «Вероятная природа внутренней симметрии кристаллов». Природа. 29 (748): 404. Bibcode:1884Натура..29..404Б. Дои:10.1038 / 029404b0. S2CID 4016086.
- ^ Эйнштейн А (1905). "Uber einen die Erzeugung und Verwandlung des Lichtes betreffenden heuristischen Gesichtspunkt" [Эвристическая модель создания и трансформации света]. Annalen der Physik (на немецком). 17 (6): 132. Bibcode:1905AnP ... 322..132E. Дои:10.1002 / andp.19053220607.. An английский перевод доступен из Wikisource.
- ^ Сравнивать: Эйнштейн А (1909). "Uber die Entwicklung unserer Anschauungen über das Wesen und die Konstitution der Strahlung" [Развитие наших взглядов на состав и сущность излучения]. Physikalische Zeitschrift (на немецком). 10: 817.. An английский перевод доступен из Wikisource.
- ^ Паис А (1982). Тонкость - это Господь: наука и жизнь Альберта Эйнштейна. Oxford University Press. ISBN 0-19-853907-X.
- ^ Комптон А (1923). «Квантовая теория рассеяния рентгеновских лучей на элементах света» (PDF). Phys. Rev. 21 (5): 483. Bibcode:1923ПхРв ... 21..483С. Дои:10.1103 / PhysRev.21.483.
- ^ Брэгг WH (1907). «Природа рентгеновских лучей». Труды Королевского научного общества Австралии. 31: 94.
- ^ Брэгг WH (1908). «Природа γ- и рентгеновских лучей». Природа. 77 (1995): 270. Bibcode:1908Натура..77..270Б. Дои:10.1038 / 077270a0. S2CID 4020075. Смотрите также Брэгг, У. Х. (1908). «Природа γ и рентгеновских лучей». Природа. 78 (2021): 271. Bibcode:1908Натура..78..271Б. Дои:10.1038 / 078271a0. S2CID 4039315. Брэгг, В. Х. (1908). «Природа γ- и рентгеновского излучения». Природа. 78 (2022): 293. Bibcode:1908Натура..78..293Б. Дои:10.1038 / 078293d0. S2CID 3993814. Брэгг, У. Х. (1908). «Природа рентгеновских лучей». Природа. 78 (2035): 665. Bibcode:1908Натура..78Р.665Б. Дои:10.1038 / 078665b0. S2CID 4024851.
- ^ Брэгг WH (1910). «Последствия корпускулярной гипотезы γ- и рентгеновских лучей и диапазона β-лучей». Фил. Mag. 20 (117): 385. Дои:10.1080/14786441008636917.
- ^ Брэгг WH (1912). «О прямом или косвенном характере ионизации рентгеновскими лучами». Фил. Mag. 23 (136): 647. Дои:10.1080/14786440408637253.
- ^ а б Фридрих В; Knipping P; фон Лауэ М. (1912). "Interferenz-Erscheinungen bei Röntgenstrahlen". Sitzungsberichte der Mathematisch-Physikalischen Classe der Königlich-Bayerischen Akademie der Wissenschaften zu München. 1912: 303.
- ^ фон Лауэ М. (1914). «Об обнаружении рентгеновских помех» (PDF). Нобелевские лекции по физике. 1901–1921. Получено 2009-02-18.
- ^ Dana ES; Форд МЫ (1932). Учебник минералогии (четвертое изд.). Нью-Йорк: Джон Вили и сыновья. п. 28.
- ^ Андре Гинье (1952). Рентгеновская кристаллографическая технология. Лондон: Hilger and Watts LTD. п. 271.
- ^ Брэгг WL (1912). «Зеркальное отражение рентгеновских лучей». Природа. 90 (2250): 410. Bibcode:1912Натура..90..410Б. Дои:10.1038 / 090410b0. S2CID 3952319.
- ^ Брэгг WL (1913). «Дифракция коротких электромагнитных волн на кристалле». Труды Кембриджского философского общества. 17: 43.
- ^ Брэгг (1914). "Die Reflexion der Röntgenstrahlen". Jahrbuch der Radioaktivität und Elektronik. 11: 350.
- ^ Брэгг (1913). «Структура некоторых кристаллов по результатам их дифракции рентгеновских лучей». Proc. R. Soc. Лондон. A89 (610): 248–277. Bibcode:1913RSPSA..89..248B. Дои:10.1098 / rspa.1913.0083. JSTOR 93488.
- ^ Брэгг В.Л .; Джеймс РУ; Босанкет СН (1921). «Интенсивность отражения рентгеновских лучей каменной солью». Фил. Mag. 41 (243): 309. Дои:10.1080/14786442108636225.
- ^ Брэгг В.Л .; Джеймс РУ; Босанкет СН (1921). «Интенсивность отражения рентгеновских лучей каменной солью. Часть II». Фил. Mag. 42 (247): 1. Дои:10.1080/14786442108633730.
- ^ Брэгг WL; Джеймс РУ; Босанкет СН (1922). «Распределение электронов вокруг ядра в атомах натрия и хлора». Фил. Mag. 44 (261): 433. Дои:10.1080/14786440908565188.
- ^ а б Брэгг WH; Брэгг WL (1913). «Структура алмаза». Природа. 91 (2283): 557. Bibcode:1913Натура..91..557Б. Дои:10.1038 / 091557a0. S2CID 3987932.
- ^ Брэгг WH; Брэгг WL (1913). «Структура алмаза». Proc. R. Soc. Лондон. A89 (610): 277. Bibcode:1913RSPSA..89..277B. Дои:10.1098 / RSPA.1913.0084.
- ^ Брэгг WL (1914). «Кристаллическая структура меди». Фил. Mag. 28 (165): 355. Дои:10.1080/14786440908635219.
- ^ а б Брэгг WL (1914). «Анализ кристаллов рентгеновским спектрометром». Proc. R. Soc. Лондон. A89 (613): 468. Bibcode:1914RSPSA..89..468B. Дои:10.1098 / RSPA.1914.0015.
- ^ Брэгг WH (1915). «Строение шпинели группы кристаллов». Фил. Mag. 30 (176): 305. Дои:10.1080/14786440808635400.
- ^ Нисикава С (1915). «Строение некоторых кристаллов группы шпинелей». Proc. Tokyo Math. Phys. Soc. 8: 199.
- ^ Вегард Л. (1916). «Результаты анализа кристаллов». Фил. Mag. 32 (187): 65. Дои:10.1080/14786441608635544.
- ^ Аминов Г. (1919). «Кристаллическая структура пирохроита». Stockholm Geol. Fören. Для ч. 41: 407. Дои:10.1080/11035891909447000.
- ^ Аминов Г (1921). "Uber die Struktur des Magnesiumhydroxids". З. Кристаллогр. 56: 505.
- ^ Брэгг WL (1920). «Кристаллическая структура оксида цинка». Фил. Mag. 39 (234): 647. Дои:10.1080/14786440608636079.
- ^ Дебие П.; Шеррер П. (1916). "Interferenz an regellos orientierten Teilchen im Röntgenlicht I". Physikalische Zeitschrift. 17: 277.
- ^ Фридрих W (1913). "Eine neue Interferenzerscheinung bei Röntgenstrahlen". Physikalische Zeitschrift. 14: 317.
- ^ Корпус AW (1917). «Новый метод рентгеновского анализа кристаллов». Phys. Rev. 10 (6): 661. Bibcode:1917ПхРв ... 10..661Ч. Дои:10.1103 / PhysRev.10.661.
- ^ Бернал JD (1924). «Структура графита». Proc. R. Soc. Лондон. A106 (740): 749–773. JSTOR 94336.
- ^ Hassel O; Мак H (1924). "Uber die Kristallstruktur des Graphits". Zeitschrift für Physik. 25 (1): 317. Bibcode:1924ZPhy ... 25..317H. Дои:10.1007 / BF01327534. S2CID 121157442.
- ^ Корпус AW (1917 г.). «Кристаллическая структура железа». Phys. Rev. 9 (1): 84. Bibcode:1917ПхРв .... 9 ... 83.. Дои:10.1103 / PhysRev.9.83.
- ^ Халл AW (июль 1917 г.). «Кристаллическая структура магния». Труды Национальной академии наук Соединенных Штатов Америки. 3 (7): 470–3. Bibcode:1917ПНАС .... 3..470Н. Дои:10.1073 / pnas.3.7.470. ЧВК 1091290. PMID 16576242.
- ^ а б «От атомов к узорам». Коллекция Wellcome. Архивировано из оригинал 7 сентября 2013 г.. Получено 17 октября 2013.
- ^ Wyckoff RWG; Посняк Э. (1921). «Кристаллическая структура хлороплатината аммония». Варенье. Chem. Soc. 43 (11): 2292. Дои:10.1021 / ja01444a002.
- ^ а б Брэгг WH (1921). «Строение органических кристаллов». Proc. R. Soc. Лондон. 34 (1): 33. Bibcode:1921PPSL ... 34 ... 33B. Дои:10.1088/1478-7814/34/1/306.
- ^ Lonsdale K (1928). «Строение бензольного кольца». Природа. 122 (3082): 810. Bibcode:1928Натура.122..810Л. Дои:10.1038 / 122810c0. S2CID 4105837.
- ^ Полинг Л (1960). Природа химической связи (3-е изд.). Итака, штат Нью-Йорк: Издательство Корнельского университета. ISBN 0-8014-0333-2.
- ^ Брэгг WH (1922). «Кристаллическая структура антрацена». Proc. R. Soc. Лондон. 35 (1): 167. Bibcode:1922PPSL ... 35..167B. Дои:10.1088/1478-7814/35/1/320.
- ^ Пауэлл Х.М.; Юэнс Р.В.Г. (1939). «Кристаллическая структура эннеакарбонила железа». J. Chem. Soc.: 286. Дои:10.1039 / jr9390000286.
- ^ Bertrand JA; Хлопок; Dollase (1963). «Связанный металл-металл полиядерный комплексный анион в CsReCl4". Варенье. Chem. Soc. 85 (9): 1349. Дои:10.1021 / ja00892a029.
- ^ Робинсон WT; Fergusson JE; Пенфолд BR (1963). «Конфигурация аниона в CsReCl.4". Труды Лондонского химического общества: 116.
- ^ Хлопок FA, Curtis NF, Harris CB, Johnson BF, Lippard SJ, Mague JT и др. (Сентябрь 1964 г.). «Мононуклеарная и полиядерная химия рения (III): его выраженная гомофильность». Наука. 145 (3638): 1305–7. Bibcode:1964Sci ... 145.1305C. Дои:10.1126 / science.145.3638.1305. PMID 17802015. S2CID 29700317.
- ^ Хлопок FA; Харрис (1965). «Кристаллическая и молекулярная структура дигидрата октахлородирхената (III) калия». Неорганическая химия. 4 (3): 330. Дои:10.1021 / ic50025a015.
- ^ Хлопок FA (1965). "Соединение металл-металл в [Re2Икс8]2− Ионы и другие кластеры атомов металлов ». Неорганическая химия. 4 (3): 334. Дои:10.1021 / ic50025a016.
- ^ Эберхардт WH; Кроуфорд У. мл .; Липскомб WN (1954). «Валентная структура гидридов бора». J. Chem. Phys. 22 (6): 989. Bibcode:1954ЖЧФ..22..989Э. Дои:10.1063/1.1740320.
- ^ Мартин Т.В., Деревенда З.С. (май 1999 г.). «Название облигация - Н облигация». Структурная биология природы. 6 (5): 403–6. Дои:10.1038/8195. PMID 10331860. S2CID 27195273.
- ^ Dunitz JD; Orgel LE; Рич А. (1956). «Кристаллическая структура ферроцена». Acta Crystallographica. 9 (4): 373. Дои:10.1107 / S0365110X56001091.
- ^ Seiler P; Дуниц Дж. Д. (1979). «Новая интерпретация неупорядоченной кристаллической структуры ферроцена». Acta Crystallographica B. 35 (5): 1068. Дои:10.1107 / S0567740879005598.
- ^ Wunderlich JA; Меллор Д. П. (1954). «Заметка о кристаллической структуре соли Цейзе». Acta Crystallographica. 7: 130. Дои:10.1107 / S0365110X5400028X.
- ^ Джарвис Джэй; Килборн БТ; Owston PG (1970). "Повторное определение кристаллической и молекулярной структуры соли Цейзе, KPtCl.3.C2ЧАС4.ЧАС2О. Поправка ». Acta Crystallographica B. 26 (6): 876. Дои:10.1107 / S056774087000328X.
- ^ Джарвис ДжейДжей; Килборн БТ; Owston PG (1971). "Повторное определение кристаллической и молекулярной структуры соли Цейзе, KPtCl.3.C2ЧАС4.ЧАС2O ". Acta Crystallographica B. 27 (2): 366. Дои:10.1107 / S0567740871002231.
- ^ Любовь РА; Koetzle TF; Williams GJB; Andrews LC; Бау Р. (1975). «Нейтронографическое исследование структуры соли Цейзе, KPtCl.3(C2ЧАС4).ЧАС2О ". Неорганическая химия. 14 (11): 2653. Дои:10.1021 / ic50153a012.
- ^ Хэвекер, Майкл; Врабец, Сабина; Крёнерт, Ютта; Чепеи, Ленард-Иштван; Науманн д'Алнонкур, Рауль; Коленько, Юрий В .; Girgsdies, Франк; Шлёгль, Роберт; Траншке, Аннетт (2012). «Химия поверхности фазово-чистого оксида M1 MoVTeNb при работе с селективным окислением пропана до акриловой кислоты». Журнал катализа. 285: 48–60. Дои:10.1016 / j.jcat.2011.09.012. HDL:11858 / 00-001M-0000-0013-FB1F-C.
- ^ Кинетические исследования окисления пропана на смешанных оксидных катализаторах на основе Mo и V. 2011.
- ^ Науманн д'Алнонкур, Рауль; Чепеи, Ленард-Иштван; Хэвекер, Майкл; Girgsdies, Франк; Schuster, Manfred E .; Шлёгль, Роберт; Траншке, Аннетт (2014). «Реакционная сеть при окислении пропана на фазово-чистых оксидных катализаторах MoVTeNb M1». Журнал катализа. 311: 369–385. Дои:10.1016 / j.jcat.2013.12.008. HDL:11858 / 00-001M-0000-0014-F436-1.
- ^ а б Браун, Дуэйн (30 октября 2012 г.). «Первые исследования почвы марсохода NASA помогают отпечаткам пальцев марсианских минералов». НАСА. Получено 31 октября, 2012.
- ^ Вестгрен А; Фрагмен Г (1925). «Рентгеновский анализ сплавов Cu-Zn, Ag-Zn и Au-Zn». Фил. Mag. 50: 311. Дои:10.1080/14786442508634742.
- ^ Брэдли Эй Джей; Тьюлис Дж (1926). «Структура γ-латуни». Proc. R. Soc. Лондон. 112 (762): 678. Bibcode:1926RSPSA.112..678B. Дои:10.1098 / rspa.1926.0134.
- ^ Хьюм-Ротери В. (1926). «Исследования природы, свойств и условий образования интерметаллических соединений (с особым упором на некоторые соединения олова)». Журнал Института металлов. 35: 295.
- ^ Брэдли Эй Джей; Грегори CH (1927). «Структура некоторых тройных сплавов». Природа. 120 (3027): 678. Bibcode:1927Натура.120..678.. Дои:10.1038 / 120678a0.
- ^ Вестгрен А. (1932). "Zur Chemie der Legierungen". Angewandte Chemie. 45 (2): 33. Дои:10.1002 / ange.19320450202.
- ^ Бернал JD (1935). «Электронная теория металлов». Годовые отчеты об успехах химии. 32: 181. Дои:10.1039 / AR9353200181.
- ^ Полинг Л. (1923). «Кристаллическая структура станнида магния». Варенье. Chem. Soc. 45 (12): 2777. Дои:10.1021 / ja01665a001.
- ^ Полинг Л. (1929). «Принципы определения структуры сложных ионных кристаллов». Варенье. Chem. Soc. 51 (4): 1010. Дои:10.1021 / ja01379a006.
- ^ Дикинсон Р.Г .; Раймонд А.Л. (1923). «Кристаллическая структура гексаметилен-тетрамина» (PDF). Варенье. Chem. Soc. 45: 22. Дои:10.1021 / ja01654a003.
- ^ Мюллер А (1923). «Рентгенологическое исследование жирных кислот». Журнал химического общества. 123: 2043. Дои:10.1039 / ct9232302043.
- ^ Saville WB; Ширер Г (1925). «Рентгеновское исследование насыщенных алифатических кетонов». Журнал химического общества. 127: 591. Дои:10.1039 / ct9252700591.
- ^ Брэгг WH (1925). «Исследование тонких пленок с помощью рентгеновских лучей». Природа. 115 (2886): 266. Bibcode:1925 г.Натура.115..266Б. Дои:10.1038 / 115266a0.
- ^ де Бройль М; Триллат Дж. Дж. (1925). "Sur l'interprétation Physique des Spectres X d'acides gras". Comptes rendus hebdomadaires des séances de l'Académie des Sciences. 180: 1485.
- ^ Триллат Дж. Дж. (1926). "Rayons X et Composeés Organiques à longe chaine. Исследования спектрографических методов с учетом структур и ориентации". Annales de Physique. 6: 5. Дои:10.1051 / anphys / 192610060005.
- ^ Каспари В.А. (1928). «Кристаллография алифатических дикарбоновых кислот». Журнал химического общества. ?: 3235. Дои:10.1039 / jr9280003235.
- ^ Мюллер А (1928). «Рентгеновское исследование соединений с длинной цепью (н. Углеводороды)». Proc. R. Soc. Лондон. 120 (785): 437. Bibcode:1928RSPSA.120..437M. Дои:10.1098 / rspa.1928.0158.
- ^ Пайпер Ш. (1929). «Некоторые примеры информации, получаемой с помощью длинных интервалов жирных кислот». Труды общества Фарадея. 25: 348. Дои:10.1039 / tf9292500348.
- ^ Мюллер А (1929). «Связь между зигзагообразной структурой углеводородной цепи и чередованием свойств нечетных и четных соединений цепей». Proc. R. Soc. Лондон. 124 (794): 317. Bibcode:1929RSPSA.124..317M. Дои:10.1098 / rspa.1929.0117.
- ^ Робертсон Дж. М. (1936). «Рентгеновское исследование фталоцианинов, часть II». Журнал химического общества: 1195. Дои:10.1039 / jr9360001195.
- ^ Кроуфут Ходжкин Д (1935). «Рентгеновские снимки монокристаллов инсулина». Природа. 135 (3415): 591. Bibcode:1935Натура.135..591С. Дои:10.1038 / 135591a0. S2CID 4121225.
- ^ Кендрю JC, Бодо Г., Динцис Х.М., Пэрриш Р.Г., Вайкофф Х., Филипс, округ Колумбия (март 1958 г.). «Трехмерная модель молекулы миоглобина, полученная методом рентгеновского анализа». Природа. 181 (4610): 662–6. Bibcode:1958Натура.181..662K. Дои:10.1038 / 181662a0. PMID 13517261. S2CID 4162786.
- ^ "Нобелевская премия по химии 1962 г.". www.nobelprize.org. Получено 2018-01-31.
- ^ «Таблица записей в PDB, составленная экспериментальным методом».
- ^ «Статистика PDB». Банк данных белков RCSB. Получено 2010-02-09.
- ^ Скапин Г (2006). «Структурная биология и открытие лекарств». Текущий фармацевтический дизайн. 12 (17): 2087–97. Дои:10.2174/138161206777585201. PMID 16796557.
- ^ Lundstrom K (ноябрь 2006 г.). «Структурная геномика мембранных белков». Клеточные и молекулярные науки о жизни. 63 (22): 2597–607. Дои:10.1007 / s00018-006-6252-у. PMID 17013556. S2CID 13432321.
- ^ Lundstrom K (август 2004 г.). «Структурная геномика мембранных белков: мини-обзор». Комбинаторная химия и высокопроизводительный скрининг. 7 (5): 431–9. Дои:10.2174/1386207043328634. PMID 15320710.
- ^ Чинте У, Шах Б., Чен Ю.С., Пинкертон А.А., Шалль, Калифорния, Хэнсон Б.Л. (апрель 2007 г.). «Криогенное (<20 K) гелиевое охлаждение снижает радиационное повреждение кристаллов белка». Acta Crystallographica. Раздел D, Биологическая кристаллография. 63 (Pt 4): 486–92. Дои:10.1107 / s0907444907005264. PMID 17372353.
- ^ Claydon, J .; Гривз, Н. и Уоррен, С. (2012). Органическая химия (PDF) (2-е изд.). Издательство Оксфордского университета. п. 45. ISBN 978-0199270293.CS1 maint: несколько имен: список авторов (связь)
- ^ Баскаран К., Дуарте Дж. М., Бияни Н., Бливен С., Капитани Дж. (Октябрь 2014 г.). «Оценка взаимодействия белок-белок в рамках всего PDB, основанная на эволюции». BMC Структурная биология. 14 (1): 22. Дои:10.1186 / s12900-014-0022-0. ЧВК 4274722. PMID 25326082.
- ^ Леви Э.Д. (ноябрь 2007 г.). «PiQSi: исследование четвертичной структуры белков». Структура. 15 (11): 1364–7. Дои:10.1016 / j.str.2007.09.019. PMID 17997962.
- ^ Сурьянараяна, Ч .; Нортон, М. Грант (29.06.2013). Рентгеновская дифракция: практический подход. Springer Science & Business Media. ISBN 9781489901484.
- ^ Грейлингер, Олден Б. (1935). "Метод Лауэ обратного отражения для определения ориентации кристаллов". Zeitschrift für Kristallographie - Кристаллические материалы. 91 (1–6). Дои:10.1524 / zkri.1935.91.1.424. S2CID 101434745.
- ^ Аналогичную дифракционную картину можно наблюдать, направив лазерную указку на компакт-диск или же DVD; периодическое расположение дорожек компакт-диска соответствует периодическому расположению атомов в кристалле.
- ^ "Морфологический XRD анализ | IMR TEST LABS". www.imrtest.com. Получено 2018-04-30.
- ^ Джонс Н. (январь 2014 г.). «Кристаллография: атомные секреты». Природа. 505 (7485): 602–3. Bibcode:2014Натура.505..602J. Дои:10.1038 / 505602a. PMID 24476871.
- ^ Мяо, Цзяньвэй; Хараламбус, Памбос; Кирз, Янош; Сейр, Дэвид (1999). «Расширение методологии рентгеновской кристаллографии для получения изображений некристаллических образцов микрометрового размера». Природа. 400 (6742): 342. Bibcode:1999Натура 400..342М. Дои:10.1038/22498. S2CID 4327928.
- ^ Харп Дж. М., Тимм Д. Е., Буник Дж. Дж. (Июль 1998 г.). «Отжиг макромолекулярных кристаллов: преодоление повышенной мозаичности, связанной с криокристаллографией». Acta Crystallographica. Раздел D, Биологическая кристаллография. 54 (Пт 4): 622–8. Дои:10.1107 / S0907444997019008. PMID 9761858.
- ^ Харп Дж. М., Хэнсон Б. Л., Тимм Д. Е., Буник Дж. Дж. (Июль 1999 г.). «Отжиг макромолекулярных кристаллов: оценка методов и переменных». Acta Crystallographica. Раздел D, Биологическая кристаллография. 55 (Pt 7): 1329–34. Дои:10.1107 / S0907444999005442. PMID 10393299.
- ^ Хэнсон Б.Л., Харп Дж. М., Буник Г. Дж. (2003). «Закаленный кристалл протеина: макромолекулярные кристаллы отжига». Макромолекулярная кристаллография, часть C. Методы в энзимологии. 368. С. 217–35. Дои:10.1016 / S0076-6879 (03) 68012-2. ISBN 978-0-12-182271-2. PMID 14674276.
- ^ Герлоф А., Браун Дж., Кутар Б., Эглофф М.П., Энгита Ф.Дж., Фогг М.Дж. и др. (Октябрь 2006 г.). «Влияние характеристики белков на структурную протеомику». Acta Crystallographica. Раздел D, Биологическая кристаллография. 62 (Пт 10): 1125–36. Дои:10.1107 / S0907444906030307. ЧВК 7161605. PMID 17001090.
- ^ Чернов А.А. (апрель 2003 г.). «Кристаллы белка и их рост». Журнал структурной биологии. 142 (1): 3–21. Дои:10.1016 / S1047-8477 (03) 00034-0. PMID 12718915.
- ^ Тереза Бергфорс (2016). "Учебное пособие по кристаллизации белков".
- ^ Чайен, Наоми. «Ограничения кристаллизации под маслом». Получено 2 апреля 2019. Цитировать журнал требует
| журнал =(помощь) - ^ Рупп Б., Ван Дж. (Ноябрь 2004 г.). «Прогностические модели кристаллизации белков». Методы. 34 (3): 390–407. Дои:10.1016 / j.ymeth.2004.03.031. PMID 15325656.
- ^ Чайен NE (июль 2005 г.). «Способы разделения зародышеобразования и роста при кристаллизации белка». Прогресс в биофизике и молекулярной биологии. 88 (3): 329–37. Дои:10.1016 / j.pbiomolbio.2004.07.007. PMID 15652248.
- ^ Сток Д., Перишич О., Лёве Дж. (Июль 2005 г.). «Роботизированная кристаллизация нанолитрового белка в лаборатории молекулярной биологии MRC». Прогресс в биофизике и молекулярной биологии. 88 (3): 311–27. Дои:10.1016 / j.pbiomolbio.2004.07.009. PMID 15652247.
- ^ Джерузалми Д. (2006). «Первый анализ макромолекулярных кристаллов: биохимия и дифракция рентгеновских лучей». Протоколы кристаллографии макромолекул, том 2. Методы молекулярной биологии. 364. С. 43–62. Дои:10.1385/1-59745-266-1:43. ISBN 1-59745-266-1. PMID 17172760.
- ^ Хелливелл-младший (июнь 2005 г.). «Совершенный протеиновый кристалл и его применение». Acta Crystallographica. Раздел D, Биологическая кристаллография. 61 (Pt 6): 793–8. Дои:10.1107 / S0907444905001368. PMID 15930642.
- ^ Ванденберг Дж.М., Темкин Х., Хамм Р.А., ДиДжузеппе М.А. (1982). «Структурное исследование контактов металлизации из легированного золота на слоях InGaAsP / InP». Журнал прикладной физики. 53 (11): 7385–7389. Bibcode:1982JAP .... 53.7385V. Дои:10.1063/1.330364.
- ^ Ванденберг JM, Темкин H (1984). «Рентгеновское исследование взаимодействия золота / барьерного металла со слоями InGaAsP / InP на месте». Журнал прикладной физики. 55 (10): 3676–3681. Bibcode:1984JAP .... 55.3676V. Дои:10.1063/1.332918.
- ^ Гарман, Э. Ф .; Шнайдер, Т. Р. (1997). «Макромолекулярная криокристаллография». Журнал прикладной кристаллографии. 30 (3): 211. Дои:10.1107 / S0021889897002677.
- ^ Schlichting I, Miao J (октябрь 2012 г.). «Новые возможности структурной биологии с рентгеновскими лазерами на свободных электронах». Текущее мнение в структурной биологии. 22 (5): 613–26. Дои:10.1016 / j.sbi.2012.07.015. ЧВК 3495068. PMID 22922042.
- ^ Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J (август 2000 г.). «Возможности для визуализации биомолекул с фемтосекундными импульсами рентгеновского излучения». Природа. 406 (6797): 752–7. Bibcode:2000Натура406..752Н. Дои:10.1038/35021099. PMID 10963603. S2CID 4300920.
- ^ Лю В., Вакер Д., Гати С., Хан Г. В., Джеймс Д., Ван Д. и др. (Декабрь 2013). «Последовательная фемтосекундная кристаллография рецепторов, связанных с G-белком». Наука. 342 (6165): 1521–4. Bibcode:2013Научный ... 342.1521Л. Дои:10.1126 / science.1244142. ЧВК 3902108. PMID 24357322.
- ^ Равелли РБ, Гарман Э.Ф. (октябрь 2006 г.). «Радиационные повреждения в макромолекулярной криокристаллографии». Текущее мнение в структурной биологии. 16 (5): 624–9. Дои:10.1016 / j.sbi.2006.08.001. PMID 16938450.
- ^ Пауэлл HR (октябрь 1999 г.). "Алгоритм автоиндексирования Фурье Россмана в MOSFLM". Acta Crystallographica. Раздел D, Биологическая кристаллография. 55 (Pt 10): 1690–5. Дои:10.1107 / S0907444999009506. PMID 10531518.
- ^ Hauptman H (октябрь 1997 г.). «Методы фазирования в кристаллографии белков». Текущее мнение в структурной биологии. 7 (5): 672–80. Дои:10.1016 / S0959-440X (97) 80077-2. PMID 9345626.
- ^ Усон I, Шелдрик GM (октябрь 1999 г.). «Достижения прямых методов кристаллографии белков». Текущее мнение в структурной биологии. 9 (5): 643–8. Дои:10.1016 / S0959-440X (99) 00020-2. PMID 10508770.
- ^ а б Тейлор Дж. (Ноябрь 2003 г.). «Фазовая проблема». Acta Crystallographica. Раздел D, Биологическая кристаллография. 59 (Pt 11): 1881–90. Дои:10.1107 / S0907444903017815. PMID 14573942.
- ^ Ealick SE (октябрь 2000 г.). "Достижения в области кристаллографии аномальной дифракции с множеством длин волн". Современное мнение в области химической биологии. 4 (5): 495–9. Дои:10.1016 / S1367-5931 (00) 00122-8. PMID 11006535.
- ^ Из файла PDB 2NRL, остатки 17–32.
- ^ http://proteopedia.org/wiki/index.php/Garman_lab:_Interconversion_of_lysosomal_enzyme_specificities, получено 28.11.2018.
- ^ Паркин Г (1993). «Связь-растяжение изомерии в комплексах переходных металлов: переоценка кристаллографических данных». Chem. Rev. 93 (3): 887–911. Дои:10.1021 / cr00019a003.
- ^ Ритвельд, Х. М. (1969-06-02). «Метод уточнения профиля ядерных и магнитных структур». Журнал прикладной кристаллографии. 2 (2): 65–71. Дои:10.1107 / S0021889869006558.
- ^ Метод Ритвельда. Янг, Р. А. (Роберт Алан), 1921-. [Честер, Англия]: Международный союз кристаллографии. 1993 г. ISBN 0198555776. OCLC 26299196.CS1 maint: другие (связь)
- ^ «МСКр». www.iucr.org. Получено 2019-04-06.
- ^ «Фулпроф». www.ill.eu. Получено 2019-04-06.
- ^ Петржичек, Вацлав; Душек, Михал; Палатинус, Лукаш (01.01.2014). «Кристаллографическая вычислительная система JANA2006: Общие характеристики». Zeitschrift für Kristallographie - Кристаллические материалы. 229 (5). Дои:10.1515 / zkri-2014-1737. ISSN 2196-7105. S2CID 101692863.
- ^ Луттеротти, Лука (февраль 2010 г.). «Полная подгонка рисунка для комбинированного определения размера-деформации-напряжения-текстуры при дифракции тонких пленок». Ядерные инструменты и методы в физических исследованиях Секция B: Взаимодействие пучка с материалами и атомами. 268 (3–4): 334–340. Bibcode:2010НИМПБ.268..334Л. Дои:10.1016 / j.nimb.2009.09.053. ISSN 0168-583X.
- ^ Lutterotti, L .; Бортолотти, М .; Искья, G .; Lonardelli, I .; Венк, Х.-Р. (2007), «Анализ текстуры Ритвельда по дифракционным изображениям», Десятая Европейская конференция по порошковой дифракции, ОЛЬДЕНБУРГ ВИССЕНШАФТСВЕРЛАГ, Дои:10.1524/9783486992540-020, ISBN 9783486992540
- ^ Lutterotti, L .; Matthies, S .; Wenk, H.-R .; Schultz, A. S .; Ричардсон, Дж. У. (15 января 1997 г.). «Комбинированный анализ текстуры и структуры деформированного известняка по спектрам дифракции нейтронов по времени пролета». Журнал прикладной физики. 81 (2): 594–600. Bibcode:1997JAP .... 81..594L. Дои:10.1063/1.364220. ISSN 0021-8979.
- ^ «Дистрибутивные файлы для пакета RIETAN-FP-VENUS». fujioizumi.verse.jp. Получено 2019-04-06.
- ^ Тоби, Брайан Х .; Фон Дриле, Роберт Б. (14 марта 2013 г.). «GSAS-II: генезис современного универсального программного пакета для кристаллографии с открытым исходным кодом». Журнал прикладной кристаллографии. 46 (2): 544–549. Дои:10.1107 / s0021889813003531. ISSN 0021-8898.
- ^ "DIFFRAC.SUITE TOPAS - Программное обеспечение XRD, дифракция рентгеновских лучей". Bruker.com. Получено 2019-04-06.
- ^ «Матч! - Определение фаз по порошковой дифракции». www.crystalimpact.com. Получено 2019-04-06.
- ^ Касас-Кабанас, Монтсе; Рейно, Марин; Рикарте, Йокин; Горбах, Павел; Родригес-Карвахаль, Хуан (01.12.2016). «НЕИСПРАВНОСТИ: программа доработки конструкций с обширными дефектами». Журнал прикладной кристаллографии. 49 (6): 2259–2269. Дои:10.1107 / S1600576716014473. ISSN 1600-5767.
- ^ Паттерсон А. Л. (1935). «Прямой метод определения составляющих межатомных расстояний в кристаллах». Zeitschrift für Kristallographie. 90 (1–6): 517. Дои:10.1524 / zkri.1935.90.1.517. S2CID 102041995.
- ^ "Нобелевская премия по физике 1914 г.". Нобелевский фонд. Получено 2008-10-09.
- ^ а б "Нобелевская премия по физике 1915 г.". Нобелевский фонд. Получено 2008-10-09.
- ^ а б "Нобелевская премия по химии 1962 г.". Nobelprize.org. Получено 2008-10-06.
- ^ а б c "Нобелевская премия по физиологии и медицине 1962 г.". Нобелевский фонд. Получено 2007-07-28.
- ^ "Нобелевская премия по химии 1964 г.". Nobelprize.org. Получено 2008-10-06.
- ^ а б "Нобелевская премия по химии 1972 г.". Nobelprize.org. Получено 2008-10-06.
- ^ "Нобелевская премия по химии 1976 г.". Nobelprize.org. Получено 2008-10-06.
- ^ а б "Нобелевская премия по химии 1985 г.". Nobelprize.org. Получено 2008-10-06.
- ^ а б c "Нобелевская премия по химии 1988 г.". Nobelprize.org. Получено 2008-10-06.
- ^ "Нобелевская премия по химии 1997 г.". Nobelprize.org. Получено 2008-10-06.
- ^ а б «Нобелевская премия по химии 2003 г.». Nobelprize.org. Получено 2008-10-06.
- ^ «Нобелевская премия по химии 2006 г.». Nobelprize.org. Получено 2008-10-06.
- ^ а б c «Нобелевская премия по химии 2009 г.». Nobelprize.org. Получено 2009-10-07.
- ^ «Нобелевская премия по химии 2012 г.». Nobelprize.org. Получено 2012-10-13.
- ^ Макс фон Лауэ (1937). Диаграммы Лауэ. Bangalore Press. п. 9.
- ^ Франция, Андре Отье, Institut de Minéralogie et de Physique des Milieux Condensés, Université P. et M. Curie, Paris (2013). Первые дни рентгеновской кристаллографии (Первое изд.). Оксфорд: Издательство Оксфордского университета. С. 1–8. ISBN 9780199659845.
- ^ Тангадурай С., Абрахам Дж. Т., Шривастава А. К., Мурти М. Н., Шукла С. К., Анджанеюлу Ю. (июль 2005 г.). «Картины порошковой рентгеновской дифракции для некоторых бета-лактамных, тетрациклиновых и макролидных антибиотиков». Аналитические науки. 21 (7): 833–8. Дои:10.2116 / analsci.21.833. PMID 16038505.
- ^ Rendle DF (декабрь 2005 г.). «Достижения химии применительно к судебной медицине». Обзоры химического общества. 34 (12): 1021–30. Дои:10.1039 / b415890n. PMID 16284668.
- ^ Svarcová S, Kocí E, Bezdicka P, Hradil D, Hradilová J (сентябрь 2010 г.). «Оценка лабораторной порошковой микродифракции рентгеновских лучей для приложений в области культурного наследия и судебной медицины». Аналитическая и биоаналитическая химия. 398 (2): 1061–76. Дои:10.1007 / s00216-010-3980-5. PMID 20640895. S2CID 11891108.
- ^ Хиллер JC, Томпсон TJ, Evison MP, Чемберлен AT, Wess TJ (декабрь 2003 г.). «Минеральное изменение кости во время экспериментального нагревания: рассеяние рентгеновских лучей». Биоматериалы. 24 (28): 5091–7. Дои:10.1016 / s0142-9612 (03) 00427-7. PMID 14568425.
- ^ Кортленд, Рэйчел (17 марта 2017 г.). «Рентгеновская карта трехмерного интерьера интегральных схем». IEEE Spectrum: Новости технологий, инженерии и науки. Получено 27 января 2018.
дальнейшее чтение
Международные таблицы для кристаллографии
- Тео Хан, изд. (2002). Международные таблицы для кристаллографии. Том A, пространственно-групповая симметрия (5-е изд.). Дордрехт: Kluwer Academic Publishers, для Международный союз кристаллографии. ISBN 0-7923-6590-9.
- Майкл Г. Россманн; Эдди Арнольд, ред. (2001). Международные таблицы для кристаллографии. Том F, Кристаллография биологических молекул. Dordrecht: Kluwer Academic Publishers, для Международного союза кристаллографии. ISBN 0-7923-6857-6.
- Тео Хан, изд. (1996). Международные таблицы для кристаллографии. Краткое учебное издание тома А, Симметрия пространственных групп (4-е изд.). Dordrecht: Kluwer Academic Publishers, для Международного союза кристаллографии. ISBN 0-7923-4252-6.
Связанные сборники статей
- Чарльз В. Картер; Роберт М. Свит., Ред. (1997). Макромолекулярная кристаллография, часть A (методы в энзимологии, v. 276). Сан-Диего: Academic Press. ISBN 0-12-182177-3.
- Чарльз В. Картер-младший; Роберт М. Свит., Ред. (1997). Макромолекулярная кристаллография, часть B (методы в энзимологии, v. 277). Сан-Диего: Academic Press. ISBN 0-12-182178-1.
- А. Дюкрюа; R. Giegé, eds. (1999). Кристаллизация нуклеиновых кислот и белков: практический подход (2-е изд.). Оксфорд: Издательство Оксфордского университета. ISBN 0-19-963678-8.
Учебники
- Биркхольц М., при участии Фьюстера П.Ф. и Гензеля С. (2005). «Глава 1: Принципы_дифракции рентгеновских лучей». Анализ тонких пленок с помощью рассеяния рентгеновских лучей. Вайнхайм: Wiley-VCH. ISBN 978-3-527-31052-4.
- Удар D (2002). Краткое содержание кристаллографии для биологов. Оксфорд: Издательство Оксфордского университета. ISBN 0-19-851051-9.
- Burns G .; Глейзер А. М. (1990). Космические группы для ученых и инженеров (2-е изд.). Бостон: Academic Press, Inc. ISBN 0-12-145761-3.
- Клегг В. (1998). Определение кристаллической структуры (Oxford Chemistry Primer). Оксфорд: Издательство Оксфордского университета. ISBN 0-19-855901-1.
- Каллити Б.Д. (1978). Элементы рентгеновской дифракции (2-е изд.). Ридинг, Массачусетс: издательство Addison-Wesley Publishing Company. ISBN 0-534-55396-6.
- Дрент Дж. (1999). Принципы рентгеновской кристаллографии белков. Нью-Йорк: Springer-Verlag. ISBN 0-387-98587-5.
- Джаковаццо С (1992). Основы кристаллографии. Оксфорд: Издательство Оксфордского университета. ISBN 0-19-855578-4.
- Glusker JP; Льюис М; Росси М (1994). Анализ кристаллической структуры для химиков и биологов. Нью-Йорк: Издательство VCH. ISBN 0-471-18543-4.
- Масса В. (2004). Определение кристаллической структуры. Берлин: Springer. ISBN 3-540-20644-2.
- Макферсон А (1999). Кристаллизация биологических макромолекул. Колд-Спринг-Харбор, Нью-Йорк: Лаборатория Колд-Спринг-Харбор. ISBN 0-87969-617-6.
- Макферсон А (2003). Введение в кристаллографию макромолекул. Джон Вили и сыновья. ISBN 0-471-25122-4.
- Макри Д.Е. (1993). Практическая кристаллография белков. Сан-Диего: Academic Press. ISBN 0-12-486050-8.
- О'Киф М; Гайд Б. Дж. (1996). Кристаллические структуры; I. Паттерны и симметрия. Вашингтон, округ Колумбия: Минералогическое общество Америки, серия монографий. ISBN 0-939950-40-5.
- Родос G (2000). Кристаллография стала кристально чистой. Сан-Диего: Academic Press. ISBN 0-12-587072-8., PDF-копия избранных глав
- Рупп Б (2009). Биомолекулярная кристаллография: принципы, практика и применение в структурной биологии. Нью-Йорк: Наука Гарланд. ISBN 978-0-8153-4081-2.
- Уоррен Б. Э. (1969). Дифракция рентгеновских лучей. Нью-Йорк. ISBN 0-486-66317-5.
- Захариасен WH (1945). Теория дифракции рентгеновских лучей в кристаллах.. Нью-Йорк: Dover Publications. LCCN 67026967.
Прикладной вычислительный анализ данных
- Янг, Р.А., изд. (1993). Метод Ритвельда. Оксфорд: Издательство Оксфордского университета и Международный союз кристаллографии. ISBN 0-19-855577-6.
Исторический
- Bijvoet JM; Burgers WG; Hägg G; ред. (1969). Ранние работы по дифракции рентгеновских лучей на кристаллах. я. Утрехт: опубликовано для Международного союза кристаллографии Uitgeversmaatschappij N.V. A. Oosthoek.CS1 maint: дополнительный текст: список авторов (связь)
- Bijvoet JM; Burgers WG; Hägg G, ред. (1972). Ранние работы по дифракции рентгеновских лучей на кристаллах. II. Утрехт: опубликовано для Международного союза кристаллографии Uitgeversmaatschappij N.V. A. Oosthoek.
- Bragg W L; Филлипс Д. К. и Липсон Н. (1992). Развитие рентгеновского анализа. Нью-Йорк: Дувр. ISBN 0-486-67316-2.
- Эвальд, PP; и многочисленные кристаллографы, ред. (1962). Пятьдесят лет рентгеновской дифракции. Утрехт: опубликовано для Международного союза кристаллографии Uitgeversmaatschappij N.V. A. Oosthoek. Дои:10.1007/978-1-4615-9961-6. ISBN 978-1-4615-9963-0.
- Эвальд П. П., редактор 50 лет рентгеновской дифракции (Перепечатано в формате pdf для XVIII Конгресса IUCr, Глазго, Шотландия, Международный союз кристаллографии).
- Фридрих W (1922). "Die Geschichte der Auffindung der Röntgenstrahlinterferenzen". Die Naturwissenschaften. 10 (16): 363. Bibcode:1922NW ..... 10..363F. Дои:10.1007 / BF01565289. S2CID 28141506.
- Лонсдейл, К. (1949). Кристаллы и рентгеновские лучи. Нью-Йорк: Д. ван Ностранд.
- «Структуры жизни». Министерство здравоохранения и социальных служб США. 2007 г. Цитировать журнал требует
| журнал =(помощь)
внешняя ссылка
| Библиотечные ресурсы о Рентгеновская кристаллография |
Учебники
- Изучение кристаллографии
- Простое, нетехническое введение
- Коллекция кристаллографии, видеоряд от Королевский институт
- «Кристаллизация малых молекул» (PDF ) в Иллинойсский технологический институт интернет сайт
- Международный союз кристаллографии
- Кристаллография 101
- Интерактивное руководство по структурному фактору, демонстрирующие свойства дифракционной картины 2D-кристалла.
- Книга с изображениями преобразований Фурье, illustrating the relationship between crystal and diffraction pattern in 2D.
- Lecture notes on X-ray crystallography and structure determination
- Online lecture on Modern X-ray Scattering Methods for Nanoscale Materials Analysis by Richard J. Matyi
- Interactive Crystallography Timeline от Королевский институт
Первичные базы данных
- Открытая база данных кристаллографии (COD)
- Банк данных белков (PDB )
- Nucleic Acid Databank (NDB)
- Кембриджская структурная база данных (CSD )
- Inorganic Crystal Structure Database (ICSD )
- Biological Macromolecule Crystallization Database (BMCD)
Производные базы данных
- PDBsum
- Proteopedia – the collaborative, 3D encyclopedia of proteins and other molecules
- RNABase
- HIC-Up database of PDB ligands
- Структурная классификация белков база данных
- CATH Protein Structure Classification
- List of transmembrane proteins with known 3D structure
- Ориентации белков в базе данных мембран
Структурная проверка
- MolProbity structural validation suite
- ProSA-web
- NQ-Flipper (check for unfavorable rotamers of Asn and Gln residues)
- DALI server (identifies proteins similar to a given protein)