Синдром Хантера - Hunter syndrome
| Синдром Хантера | |
|---|---|
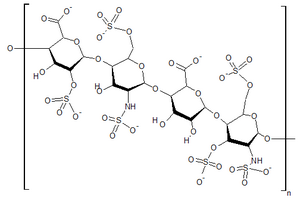 | |
| Структура гепарансульфат, один из GAGs накапливается в тканях людей с синдромом Хантера | |
| Специальность | Эндокринология |
| Симптомы | Скелетные аномалии, потеря слуха, дегенерация сетчатки, увеличение печени и селезенки |
| Осложнения | Заболевания верхних дыхательных путей; сердечно-сосудистая недостаточность |
| Причины | Дефицит фермента идуронат-2-сульфатаза |
| Дифференциальная диагностика | Мукополисахаридоз I типа; Другой мукополисахаридозы |
| Прогноз | В тяжелых случаях смерть обычно наступает к 15 годам. В более тяжелых случаях пациенты могут дожить до 50 лет. |
| Частота | 1 из 100 000–150 000 новорожденных мальчиков[1] |
Синдром Хантера, или же мукополисахаридоз тип II (MPS II), является редким генетическим заболеванием, при котором большие молекулы сахара называются гликозаминогликаны (или ГАГ или мукополисахариды) накапливаются в тканях организма. Это форма лизосомная болезнь накопления. Синдром Хантера вызван недостатком лизосомный фермент идуронат-2-сульфатаза (I2S).[2][3] Отсутствие этого фермент причины гепарансульфат и дерматансульфат накапливаться во всех тканях организма.[4] Синдром Хантера - единственный МПС синдром выставлять Х-сцепленный рецессивный наследование.[4]
Симптомы синдрома Хантера сопоставимы с симптомами MPS I. Это вызывает аномалии во многих органах, включая скелет, сердце и дыхательную систему. В тяжелых случаях это приводит к смерти в подростковом возрасте. В отличие от MPS I, помутнение роговицы не связано с этим заболеванием.[1]
Признаки и симптомы
Синдром Хантера может проявляться большим разнообразием фенотипы. Это традиционно классифицируется как «легкое» или «тяжелое» в зависимости от наличия Центральная нервная система симптомы, но это чрезмерное упрощение. Пациенты с «ослабленной» или «легкой» формой заболевания могут по-прежнему страдать от серьезных проблем со здоровьем. Для серьезно пораженных пациентов клиническое течение относительно предсказуемо; пациенты обычно умирают в раннем возрасте. Для людей с более легкими формами болезни существует более широкий спектр исходов. Многие доживают до 20-30 лет, но у некоторых средняя продолжительность жизни может быть близка к нормальной, и они могут даже иметь детей. Сердечные и респираторные нарушения - обычная причина смерти пациентов с более легкими формами болезни.[2]
Симптомы синдрома Хантера (МПС II) обычно не проявляются при рождении. Часто первые симптомы могут включать: брюшные грыжи, ушные инфекции, насморк, и простуды. По мере того, как накопление ГАГ продолжается по клеткам тела, признаки MPS II становятся более заметными. Внешность многих детей с синдромом включает отчетливую грубость в чертах лица, в том числе заметную лоб, нос с уплощенной переносицей и увеличенным язык. У них также может быть большая голова, а также увеличенный живот. В тяжелых случаях МПС II диагноз часто ставится в возрасте от 18 до 36 месяцев. В более легких случаях пациенты поступают так же, как и дети с Синдром Херлера – Шейе, и диагноз обычно ставится в возрасте от 4 до 8 лет.[2]
Продолжительное хранение ГАГ приводит к аномалиям во многих системах органов. После 18 месяцев дети с тяжелым МПС II могут страдать от замедления развития и прогрессирующей потери навыков.[1] Утолщение сердечных клапанов и стенок сердца может привести к прогрессирующему снижению сердечной функции. Стенки дыхательных путей также могут утолщаться, что приводит к обструктивная болезнь дыхательных путей. Поскольку печень и селезенка со временем становится больше, живот может стать растянутый, делая более заметными грыжи. Все основные суставы может пострадать от MPS II, что приведет к тугоподвижность суставов и ограниченное движение. Прогрессирующее поражение пальца и большого пальца суставы приводит к снижению способности подбирать мелкие предметы. Воздействие на другие суставы, такие как бедра и колени, может затруднить нормальную ходьбу. Если синдром запястного канала развивается, может произойти дальнейшее снижение функции руки. Сами кости могут быть поражены, что приведет к низкому росту. Кроме того, галечная кожа цвета слоновой кости поражения может быть обнаружен на плечах, ногах и верхней части спины некоторых людей. Эти поражения кожи считаются патогномический за болезнь. Наконец, хранение ГАГов в мозг может привести к задержке развития с последующим Интеллектуальная недееспособность и прогрессирующая потеря функции.
Возраст появления симптомов и наличие или отсутствие поведенческих расстройств являются прогностическими факторами окончательной тяжести заболевания у очень маленьких пациентов. Нарушения поведения часто могут имитировать комбинации симптомов Синдром дефицита внимания и гиперактивности, аутизм, обсессивно-компульсивное расстройство, и / или расстройство сенсорной обработки, хотя наличие и уровень симптомов различаются у каждого пораженного ребенка. Они часто также включают отсутствие надлежащего чувства опасности и агрессии. Поведенческие симптомы MPS II обычно предшествуют нейродегенерации и часто усиливаются до тех пор, пока психические расстройства не становятся более выраженными.[5] К моменту смерти большинство детей с тяжелым МПС II имеют серьезные психические отклонения и полностью зависят от своих опекунов.[2]
Генетика

Поскольку синдром Хантера является X-сцепленным рецессивным заболеванием, он преимущественно поражает пациентов мужского пола. В IDS ген расположен на Х-хромосоме. Ген IDS кодирует фермент идуронат-2-сульфатаза (I2S). Недостаток этого фермента приводит к накоплению ГАГ, которые вызывают симптомы MPS II.[6] У женщин обычно две Х-хромосомы, тогда как у мужчин обычно одна Х-хромосома, которую они наследуют от своей матери, и одну Y-хромосому, которую они наследуют от своего отца.
Если самка наследует одну копию мутанта аллель для MPS II у нее обычно будет обычная копия IDS ген, который может компенсировать мутантный аллель. Это известно как генетический носитель. Однако мужчина, унаследовавший дефектную Х-хромосому, обычно не имеет другой Х-хромосомы, компенсирующей мутантный ген. Таким образом, женщина должна унаследовать два мутантных гена для развития MPS II, тогда как пациенту мужского пола необходимо унаследовать только один мутантный ген. Женщина-носитель может пострадать из-за: X-инактивация, что является случайным процессом.[нужна цитата ]
Патофизиология
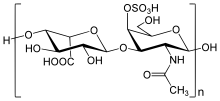
В тело человека зависит от огромного количества биохимический реакции на поддержку критических функций. Одна из таких функций - разбивка больших биомолекулы. Неудача этого процесса - основная проблема синдрома Хантера и связанных с ним нарушений памяти.
Биохимия синдрома Хантера связана с проблемой в части соединительной ткани, известной как внеклеточный матрикс, который состоит из множества сахара и белки. Помогает сформировать архитектурный каркас тела. Матрица окружает клетки тела организованной сеткой и действует как клей, который скрепляет клетки тела. Одна из частей внеклеточного матрикса - это молекула, называемая протеогликан. Как и многие компоненты организма, протеогликаны необходимо расщеплять и заменять. Когда организм расщепляет протеогликаны, одним из образующихся продуктов является мукополисахариды (ГАГ).[нужна цитата ]
В MPS II проблема касается поломки двух ГАГов: дерматансульфат и гепарансульфат. На первом этапе расщепления дерматансульфата и гепарансульфата необходим лизосомальный фермент идуронат-2-сульфатаза, или I2S. У людей с МПС II этот фермент частично или полностью неактивен. В результате ГАГ накапливаются в клетках по всему телу, особенно в тканях, которые содержат большое количество дерматансульфата и гепарансульфата. Скорость накопления ГАГ не одинакова для всех людей с MPS II, что приводит к широкому спектру медицинских проблем.[нужна цитата ]
Диагностика
Первым лабораторным скрининговым тестом на расстройство MPS является анализ мочи для ГАГов. Аномальные значения указывают на вероятность нарушения МПС. Анализ мочи может иногда быть нормальным даже если у ребенка действительно есть расстройство MPS. Окончательный диагноз MPS II ставится путем измерения активности I2S в сыворотка, белые кровяные клетки, или же фибробласты из кожи биопсия. У некоторых людей с MPS II анализ I2S ген можно определить клиническую тяжесть.
Дородовой диагностика обычно доступна путем измерения ферментативной активности I2S в амниотическая жидкость или в ворсинка хориона ткань. Если известно, что в семье присутствует конкретная мутация, можно провести пренатальное молекулярно-генетическое тестирование. Секвенирование ДНК может выявить, является ли кто-то переносчиком болезни.[2]
Уход
Из-за большого разнообразия фенотипов лечение этого расстройства определяется специально для каждого пациента. До недавнего времени не существовало эффективной терапии МПС II, поэтому паллиативная помощь использовался. Однако последние достижения привели к созданию лекарств, которые могут улучшить выживаемость и благополучие людей с МПС II.
Ферментная заместительная терапия
Идурсульфаза, очищенная форма отсутствующего лизосомального фермента, прошла клинические испытания в 2006 г.[6] и впоследствии был одобрен США Управление по контролю за продуктами и лекарствами в качестве заместительной терапии ферментами MPS II. Идурсульфаза бета, еще одно заместительное лечение фермента, было одобрено в Корее Министерство безопасности пищевых продуктов и медикаментов.
Последние достижения в заместительная ферментная терапия (ERT) с идурсульфаза Было доказано, что улучшают многие признаки и симптомы МПС II, особенно если его начать на ранней стадии заболевания. После введения его можно транспортировать в клетки для расщепления ГАГ, но, поскольку лекарство не может проникать через гематоэнцефалический барьер, не ожидается, что это приведет к улучшению когнитивных функций у пациентов с тяжелыми симптомами со стороны центральной нервной системы. Даже с помощью ФЗТ необходимо лечение различных проблем органов у самых разных медицинских специалистов.[2]
Трансплантация костного мозга и стволовых клеток
Трансплантация костного мозга и трансплантация гемопоэтических стволовых клеток (HSCT) использовались в качестве лечения в некоторых исследованиях.[7][8] Хотя трансплантация принесла пользу многим системам органов, не было показано, что она улучшает неврологические симптомы болезни. Хотя ТГСК показала себя многообещающим при лечении других расстройств МПС, его результаты пока неудовлетворительны при лечении МПС II. Было показано, что ФЗТ приводит к лучшим результатам у пациентов с МПС II.[2]
Терапия редактирования генов
В феврале 2019 года ученые-медики, работающие с Sangamo Therapeutics со штаб-квартирой в Ричмонд, Калифорния, объявила о первом "в теле" терапия редактирования генов человека навсегда изменить ДНК - у пациента с МПС II.[9] Клинические испытания Sangamo, включающие редактирование генов с использованием нуклеаза цинкового пальца продолжаются по состоянию на февраль 2019 года.[10]
Прогноз
Более раннее появление симптомов связано с худшим прогнозом. У детей с симптомами в возрасте от 2 до 4 лет смерть обычно наступает в возрасте от 15 до 20 лет. Причина смерти обычно связана с неврологическими осложнениями, обструктивным заболеванием дыхательных путей и сердечной недостаточностью. Если у пациентов минимальные неврологические нарушения, они могут дожить до 50 лет и старше.[1][6]
Эпидемиология
По оценкам, 2000 человек во всем мире страдают MPS II, 500 из которых живут в Соединенных Штатах.[11]
Исследование, проведенное в Соединенном Королевстве, показало, что заболеваемость среди мужчин составляет около одного из 130 000 живорожденных мальчиков.[12]
История
Синдром назван в честь врача Чарльза А. Хантера (1873–1955), который впервые описал его в 1917 году.[13][14]
Исследование
Начиная с 2010 г., клинические испытания фазы I / II оценивали интратекальный инъекции более концентрированной дозы идурсульфазы, чем препарат для внутривенного введения, используемый в инфузиях заместительной ферментной терапии, в надежде предотвратить снижение когнитивных функций, связанное с тяжелой формой состояния.[15] Результаты были представлены в октябре 2013 года.[16] Клинические испытания фазы II / III начались в 2014 году.[17]
В 2017 году 44-летний[18] Пациент с MPS II лечился генной терапией в попытке предотвратить дальнейшее повреждение от болезни. Это первый случай использования генной терапии. in vivo в людях.[19] В 2018 году исследование было распространено на шесть пациентов.[20]
Общество
24 июля 2004 г. Эндрю Рэгг, 38 лет, из Уортинг, Западный Сассекс, Англия, задушил подушкой своего 10-летнего сына Якова из-за инвалидности мальчика, связанной с MPS II. Военный безопасность специалист, Рэгг также утверждал, что находился в состоянии стресса после возвращения с войны в Ирак. Он отрицал убийство Иакова, но признал себя виновным в непредумышленное убийство по причине уменьшенной вместимости. Судья Энн Рафферти назвала случай «исключительным», приговорила Врагга к двум годам тюремного заключения за непредумышленное убийство, а затем на два года условно. Рафферти сказал, что "ничего не выиграет" от отправки Рэгга в тюрьму за преступление.[21][22][23]
Смотрите также
- Синдром Гурлера (MPS I )
- Синдром Санфилиппо (MPS III)
- Синдром Моркио (MPS IV)
- Пренатальное тестирование
- Генетическое консультирование
Рекомендации
- ^ а б c d «Информационный бюллетень по мукополисахаридозам». Национальный институт неврологических заболеваний и инсульта. 15 ноября 2017 г.. Получено 11 мая 2018.
- ^ а б c d е ж грамм Призрак Дж. Э., Скарпа М., Бек М. и др. (Март 2008 г.). «Мукополисахаридоз II типа (синдром Хантера): клинический обзор и рекомендации по лечению в эпоху заместительной ферментной терапии». Евро. J. Pediatr. 167 (3): 267–77. Дои:10.1007 / s00431-007-0635-4. ЧВК 2234442. PMID 18038146.
- ^ Джеймс, Уильям Д .; Бергер, Тимоти Дж .; и другие. (2006). Кожные болезни Эндрюса: клиническая дерматология. Saunders Elsevier. п. 544. ISBN 978-0-7216-2921-6.
- ^ а б Ле, Дао; Бхушан, Викас; Хофманн, Джеффри (2012). Первая помощь для USMLE, шаг 1. Макгроу-Хилл. п. 117.
- ^ Шварц, Ида В.Д. (2007). «Клиническое исследование 77 пациентов с мукополисахаридозом II типа». Acta Paediatrica. 96 (455): 63–70. Дои:10.1111 / j.1651-2227.2007.00212.x. PMID 17391446.
- ^ а б c Мюнцер, Дж; Рэйф, Дж. Э .; Бек, М; Giugliani, R; Harmatz, P; Eng, CM; Веллоди, А; Martin, R; Рамасвами, У; Gucsavas-Calikoglu, M; Виджаярагхаван, S; Wendt, S; Пуга, AC; Ульбрих, Б; Шинави, М. Клири, М; Пайпер, D; Конвей, AM; Кимура, А (август 2006 г.). «Фаза II / III клинического исследования заместительной ферментной терапии идурсульфазой при мукополисахаридозе II (синдром Хантера)». Генетика в медицине. 8 (8): 465–73. Дои:10.1097 / 01.gim.0000232477.37660.fb. PMID 16912578.
- ^ Гаффон, Н. (май 2009 г.). «Трансплантация костного мозга детям с синдромом Хантера: исход от 7 до 17 лет». 154 (5). Журнал педиатрии. С. 733–737. Дои:10.1016 / j.jpeds.2008.11.041. PMID 19167723.
- ^ Аннибали, Р. (октябрь 2013 г.). «Синдром Хантера (мукополисахаридоз типа II), тяжелый фенотип: долгосрочное наблюдение за пациентами, перенесшими трансплантацию гемопоэтических стволовых клеток». 65 (5). Минерва Педиатрия. С. 487–496. PMID 24056375.
- ^ Маркионе, Мэрилин (7 февраля 2019 г.). «Тесты предполагают, что ученые достигли 1-го места в редактировании генов« в теле »». AP Новости. Получено 7 февраля 2019.
- ^ Персонал (2 февраля 2019 г.). «Исследование возрастающей дозы редактирования генома с помощью нуклеазы цинкового пальца (ZFN), терапевтической SB-913 у субъектов с MPS II». ClinicalTrials.gov. Национальная медицинская библиотека США. Получено 7 февраля 2019.
- ^ LaTercera.com (на испанском языке)[постоянная мертвая ссылка ]
- ^ Молодой И.Д., Харпер П.С. (1982). «Заболеваемость синдромом Хантера». Гм. Genet. 60 (4): 391–2. Дои:10.1007 / BF00569230. PMID 6809596.
- ^ Синдром Хантера (Чарльз А. Хантер) в Кто это назвал?
- ^ Хантер, К. А. (1917). "Редкая болезнь двух братьев". Труды Королевского медицинского общества. Лондон. 10 (Секта Исследование Dis Child): 104–116. ЧВК 2018097. PMID 19979883.
- ^ «Фаза I / II, рандомизированное исследование безопасности и возрастающего диапазона доз интратекальной идурсульфазы-IT, вводимой в сочетании с внутривенной элапразой у педиатрических пациентов с синдромом Хантера и когнитивными нарушениями». Clinicaltrials.gov. Национальные институты здравоохранения США. 15 июня 2009 г.. Получено 22 июля 2018.
- ^ «Исследование безопасности и диапазона дозировки идурсульфазы (интратекального) введения через устройство для интратекальной доставки лекарств у педиатрических пациентов с синдромом Хантера, у которых есть поражение центральной нервной системы и которые получают лечение с помощью Elaprase® - Результаты». Clinicaltrials.gov. Национальные институты здравоохранения США. 31 октября 2013 г.. Получено 20 июля 2014.
- ^ «Исследование интратекальной идурсульфазы-IT, вводимой в сочетании с Elaprase®, у педиатрических пациентов с синдромом Хантера и ранними когнитивными нарушениями (AIM-IT)». Clinicaltrials.gov. Национальные институты здравоохранения США. Июль 2014 г.. Получено 20 июля 2014.
- ^ Маркионе, Мэрилинн (15 ноября 2017 г.). «Американские ученые пробуют редактировать первый ген в теле». Ассошиэйтед Пресс. Получено 16 ноября 2017.
- ^ Маркионе, Мэрилин (14 ноября 2017 г.). «Ученые впервые попытались отредактировать ген внутри пациента». Время. Получено 15 ноября 2017.
- ^ Маркионе, Майлинн (5 сентября 2018 г.). «Первые результаты вселяют надежду на историческую попытку редактирования генов». AP Новости. Получено 6 сентября 2018.
- ^ NEWS.BBC.co.uk, "Отец очищен от убийства сына", Новости BBC
- ^ Guardian.co.uk, "Бывший солдат САС, который задушил неизлечимо больного сына, выходит на свободу" Хранитель
- ^ NEWS.BBC.co.uk, "Обзор" прояснит законы об убийствах "" Новости BBC
внешняя ссылка
 СМИ, связанные с Синдром Хантера в Wikimedia Commons
СМИ, связанные с Синдром Хантера в Wikimedia Commons- Запись GeneReview / NIH / UW о мукополисахаридозе II типа
| Классификация | |
|---|---|
| Внешние ресурсы |