Рецептор ЛПОНП - VLDL receptor





В рецептор липопротеинов очень низкой плотности (VLDLR) это трансмембранный липопротеин рецептор семейство рецепторов липопротеинов низкой плотности (ЛПНП). VLDLR показывает значительную гомология с членами этой линии. Обнаруженный в 1992 году Т. Ямамото, VLDLR широко распространен в тканях организма, включая сердце, скелетные мышцы, жировая ткань, и мозг, но отсутствует в печени.[5] Этот рецептор играет важную роль в поглощении холестерина, метаболизме аполипопротеин E -содержащий триацилглицерин -богатые липопротеинами, и миграция нейронов в развивающемся мозге. У людей VLDLR кодируется VLDLR ген. Мутации этого гена могут привести к множеству симптомов и заболеваний, в том числе к типу I. лиссэнцефалия, гипоплазия мозжечка, и атеросклероз.
Белковая структура
VLDLR является членом семейство рецепторов липопротеинов низкой плотности (ЛПНП), который полностью состоит из типа I трансмембранный липопротеин рецепторы.
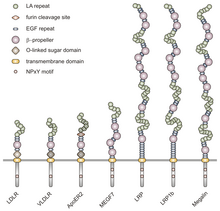
Все члены этого семейства разделяют пять высококонсервативных структурных доменов: внеклеточный N-концевой лиганд -связывающий домен с богатыми цистеином повторами (также называемыми лиганд-связывающими повторами), фактор роста эпидермиса (EGF), О-связанное гликозилирование сахарный домен, единственная трансмембранная последовательность и цитоплазматический домен, который содержит последовательность NPxY. Мотив NPxY участвует в передаче сигнала и нацеливании рецепторов на покрытые ямки и состоит из последовательности аспарагин-пролин-X-тирозин, где X может быть любой аминокислотой.[6] Имитируя эту общую структуру, VLDLR имеет восемь, 40 аминокислот длинных богатых цистеином повторов во внеклеточном N-концевом лиганд-связывающем домене.[6] Это главное отличие от основного члена семейства рецепторов ЛПНП, LDLR, который имеет всего семь богатых цистеином повторов, также состоящих из 40 аминокислот.[7] Каждый из этих богатых цистеином повторов, как в VLDLR, так и в LDLR, имеет три дисульфидные связи и координированный Ca2+ ион. N-конец также состоит из остатка глицина, за которым следуют 27 гидрофобный остатки, составляющие сигнальный пептид.[6] За этой областью следует повтор EGF, β-винт сегмент, который играет роль в pH-зависимой диссоциации комплекса лиганд-рецептор,[8] и еще два повтора EGF.[9] О-связанный домен гликозилирования VLDLR, следующий в последовательности, имеет много остатков треонина и серина и насчитывает 46 аминокислот. Трансмембранный домен, который прикрепляет рецепторы к мембране, имеет длину 22 аминокислоты.[6] Конечным в последовательности является цитоплазматический домен из 54 аминокислот, который содержит мотив NPxY.[8]
Изоформы
Полноразмерный геном VLDLR человека расположен в локусе 9p24 хромосомы 9. Он состоит из сегмента размером 40 т.п.н., который включает 19 экзон -кодирующие последовательности, которые на один экзон больше, чем кодируются LDLR. Этот дополнительный экзон в VLDLR Ген отвечает за дополнительный цистеин-связывающий повтор, не обнаруженный в LDLR.[7] Вместе экзоны, составляющие VLDLR Ген кодирует белок длиной 873 аминокислотных остатка. Известно, что VLDLR существует как четыре разных изоформы белка: тип I, II, III и IV. Эти разные изоформы являются результатом вариаций в альтернативное сращивание. Транскрипт VLDLR типа I (VLDLR-I) состоит из всех 19 экзонов. VLDLR-II, с другой стороны, не имеет экзона 16, который кодирует О-гликозилирование домен между сахарными регионами. В VLDLR-III отсутствует экзон 4, кодирующий третий лиганд -обязательный повтор. Наконец, в транскриптах VLDLR-IV отсутствуют как экзон 16, так и экзон 4. Было показано, что 75% транскриптов VLDLR существуют в виде изоформы типа II в мозг мыши модели. Это показывает, что большинство VLDLR в головном мозге не гликозилированы, поскольку у типа II отсутствует экзон 16, который кодирует домен O-гликозилирования. Известно, что изоформа IV типа является второй по значимости.[6]
Эволюционное сохранение
В пределах Семейство рецепторов ЛПНП. В частности, общая последовательность составляет 50%. гомология между VLDLR и ApoER2, еще один липопротеин рецептор этого семейства.[6] Сравнение LDLR и VLDLR было установлено, что их основные конструкции на 55% идентичны в своих лиганд -связывающие регионы. Модульные структуры этих двух белков почти наложены друг на друга, с той лишь разницей, что имеется дополнительный богатый цистеином повтор в VLDLR. Это демонстрируется выравниванием двух рецепторов в соответствии с их линкерной областью; в LDLR линкерная область расположена между богатыми цистеином повторами четырех и пяти из семи его повторов, тогда как в VLDLR линкерная область, по-видимому, находится между пятью и шестью повторами из восьми его повторов.[10]
VLDLR также демонстрирует высокую гомологию среди различных видов. ЛПОНП людей, мышей, крыс и кроликов идентифицируются на 95%. Кроме того, у цыплят сохраняется примерно 84% соответствующего белка. Этот уровень гомологии между видами намного выше, чем обнаруженный для LDLR. Следовательно, эти сравнения генов предполагают, что VLDLR и LDLR расходились раньше, чем LDLRs среди позвоночных.[10]
Связывание лиганда
VLDLR связывает соединения, содержащие аполипопротеин E (апоЕ). Эти лиганды присоединяются к повторам связывания цистеина на N-конце. Разница в богатых цистеином повторах между членами Семейство рецепторов ЛПНП приводят к различиям в аффинности связывания. VLDLR, в частности, связывает ЛПОНП и липопротеины средней плотности (IDL), но не ЛПНП. Эта неспособность связывать ЛПНП связана с неспособностью VLDLR связывать аполипопротеин B (апоВ), который присутствует в ЛПНП.[11]
Ингибиторы
Рецептор-ассоциированный белок (RAP) и тромбоспондин-1 (THBS1) были идентифицированы как соединения, которые связывают VLDLR. Во многих случаях эти соединения проявляют ингибирующие эффекты. THBS1 связывает VLDLR и блокирует связывание лиганда.[11] Это играет важную роль в катушка пути, поскольку THBS1 может блокировать прикрепление рилина, одновременно стимулируя факторы транскрипции обычно активируется reelin. Однако это связывание THBS1 не вызывает последующей деградации этих факторов транскрипции, как это делает рилин, и, таким образом, может привести к значительно усиленным эффектам.[6] Белок RAP действует аналогичным образом, блокируя связывание рилином VLDLR. Однако в этом случае также блокируется фосфорилирование факторов транскрипции, обычно осуществляемое рилином.[12]
Распределение и экспрессия тканей
VLDLR обнаружены по всему телу, с особенно высокой экспрессией в тканях жирных кислот из-за их высокого уровня триглицериды, Первичный лиганд VLDLR. Эти ткани включают ткани сердца, скелетных мышц и жировой слой. Кроме того, рецептор находится в макрофагах, эндотелиальных клетках капилляров,[8] и в головном мозге, где он выполняет совсем другие функции, чем остальные части тела. Существует предпочтительная экспрессия VLDLR типа I в сердце, скелетных мышцах и головном мозге, в отличие от типа II, который в основном экспрессируется в немышечных тканях, включая головной мозг, мозжечок, эндотелиальные клетки почек, селезенки и аорты.[7][11] Наибольшая экспрессия VLDLR обнаруживается в головном мозге. Хотя VLDLR обнаруживается почти во всех областях мозга, его максимальная экспрессия ограничена корой и мозжечком. Здесь рецептор можно найти в состоянии покоя или в активированном состоянии. микроглия которые связаны с старческие бляшки и корковые нейроны, нейробласты, ячейки матрицы, Клетки Кахаля-Ретциуса, глиобласты, астроциты, олигодендроциты, и в зависимости от региона пирамидные нейроны.[6] Несмотря на свою важную роль в метаболизме холестерина и жирных кислот, VLDLR не обнаруживается в печени. Это явление в основном объясняется очень высоким уровнем LDLR в этих областях.[7] Кроме того, было обнаружено, что этот рецептор на субклеточном уровне обнаруживается у не-липидный плот участки клеточных мембран.[6]
Регулирование
В отличие от LDLR, VLDLR не проявляет никакого механизма обратной связи и, следовательно, внутриклеточный липопротеины не могут его регулировать. Это явление связано с разницей в стерол регуляторный элемент-1 (SRE-1) VLDLR. Нормальные последовательности SRE-1, подобные тем, которые обнаруживаются в LDLR, характеризуются двумя повторами кодона CAC, разделенными двумя промежуточными нуклеотидами C (5’-CACCCCAC-3 ’). В белок, связывающий регуляторный элемент стерола -1 (SREBP-1), а фактор транскрипции, воздействует на CAC-повторы SRE-1, чтобы регулировать транскрипцию белка. Тем не менее VLDLR ген кодируется двумя последовательностями, подобными SRE-1, которые содержат однонуклеотидный полиморфизм. Эти полиморфизмы нарушают связывание SREBP-1 с повторами CAC и, следовательно, устраняют механизм обратной связи, наблюдаемый в других белках.[7]
Экспрессия VLDLR регулируется рецептор, активируемый пролифератором пероксисом, гамма (PPAR-γ). Исследование 2010 года показало, что отпускаемые по рецепту лекарства Пиоглитазон, агонист PPAR-γ увеличивает экспрессию мРНК VLDLR и уровни белка в экспериментах с использованием фибробластов мыши. Мыши, получавшие пиоглитазон, демонстрировали более высокую скорость превращения плазмы. триглицериды в эпидидимальные жиры. Как и ожидалось, мыши с дефицитом VLDLR не показали такой же реакции.[8] Эти результаты показывают, что VLDLR важны для накопления жира.[8]
Многие другие гормоны и диетические факторы также регулируют экспрессию VLDLR. Гормон щитовидной железы положительно регулирует экспрессию VLDLR в скелетных мышцах крыс, но не в жировой ткани или сердечной ткани. У кроликов экспрессия VLDLR в сердечной мышце активируется эстрогеном и подавляется эстрогеном. колониестимулирующий фактор гранулоцитов-макрофагов. В трофобласт -производных клеточных линий, повышенная экспрессия VLDLR происходит, когда клетки инкубируются с гиполипидемические средства Такие как инсулин и клофибрат. В отличие, 8-бромаденозин 3 ', 5'-циклический монофосфат (8-бром-цАМФ) подавляет экспрессию VLDLR. Наконец, на VLDLR влияет присутствие апоЕ и LDLR. Присутствие апоЕ необходимо для регуляции экспрессии VLDLR, в то время как отсутствие LDLR изменяет стерол -regulatory-element-1-подобные последовательности VLDLR, чтобы сделать их функциональными только в сердце и скелетных мышцах.[7]
Функция
За пределами нервной системы
VLDLR - периферийное липопротеин рецептор, который участвует в метаболизме липопротеинов, сердечной жирная кислота метаболизм и отложение жира. Фактически, VLDLR позволит холестерин попадать в ткани из кровотока, где он может использоваться в клеточных мембранах. Кроме того, это позволит жирным кислотам проникать в клетки, где они могут использоваться в качестве источника энергии.[7] В целом, VLDLR в первую очередь модулирует дополнительныепеченочный метаболизм триглицерид -богатые липопротеидами.[8]
Поглощение липопротеинов
VLDLR играет лишь дискретную роль в метаболизме липидов, но более значима в стрессовых ситуациях. Мыши с двойным нокауты в VLDLR и LDLR иметь более высокую сыворотку триглицерид уровней, чем те, у которых только нокаут в LDLR ген. Кроме того, LDLR Нокаутные мыши со сверхэкспрессией VLDLR имеют пониженные уровни триглицеридов в сыворотке. Хотя отложение жира близко к норме без VLDLR, его роль приобретает значение при дефиците LDLR. Несмотря на эти знания о его роли в захвате липопротеинов, полный механизм липидного метаболизма, осуществляемого VLDLR, полностью не изучен.[11]
Эндоцитоз
Известно, что VLDLR использует эндоцитоз, хотя точный механизм этого процесса для этого белка неизвестен. Эндоцитоз опосредуется последовательностями NPxY, которые, как известно, сигнализируют об интернализации рецептора через клатриновые ямы. Присутствие этой последовательности в цитоплазматическом хвосте VLDLR делает возможным эндоцитоз.[11] В целом, липопротеин рецепторы подвергаются процессу эндоцитоза со своим лигандом в ямки, покрытые клатрином. Отсюда их вместе переносят в ранние и поздние эндосомы до достижения лизосома. В этот момент происходит гидролиз, и липопротеины высвобождаются в цитоплазму, а рецепторы возвращаются обратно на поверхность клетки. Пока не подтверждено, следует ли VLDLR именно этому механизму, но вероятно один, тесно связанный с ним.[8]
В нервной системе
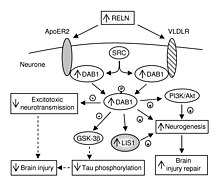
В дополнение к своей роли во всем организме, VLDLR играет уникальную роль в мозге. Это ключевой компонент катушка путь, который функционирует в миграция нейронов. VLDLR связывает белок рилина с внутриклеточным сигнальным белком, Dab1, который сообщает отдельным нейронам, куда идти в анатомии мозга. Мутации в VLDLR часто не приводят к серьезной дезорганизации, наблюдаемой в мутациях рилина. Однако мутация VLDLR действительно приводит к некоторой дезорганизации, в первую очередь локализованной в мозжечок, где VLDLR считается наиболее заметным.[6]
Нейрональная миграция
VLDLR экспрессируется на мигрирующих нейронах, помогая им найти правильное место в мозге. Этот процесс является частью катушка путь, отвечающий за формирование наизнанку шестислойной неокортекс.[6] Несмотря на открытие этого пути, многие особенности и молекулярные механизмы этого процесса все еще обсуждаются. Наличие двух рецепторов рилина, VLDLR и ApoER2, затрудняет различение конкретных функций каждого белка.[13]

VLDLR в первую очередь отвечает за правильное расположение слоев пирамидные клетки в слой 1 кора головного мозга. В частности, отсутствие VLDLR может привести к эктопическому накоплению пирамидных клеток в этой области.[13] VLDLR не влияет на миграцию ранних клеток в организованный слой, но поскольку его отсутствие приводит к вторжению этих нейробласты в маргинальную зону предполагается, что VLDLR может кодировать «стоп-сигнал». Это подтверждается тем фактом, что VLDLR в основном экспрессируется в кортикальной пластинке, прилегающей к клеткам, экспрессирующим рилин, Клетки Кахаля – Ретциуса, и в промежуточной зоне. Однако окончательных доказательств пока не найдено.[6] В целом, рилин связывает VLDLR и подвергается эндоцитоз через везикулы, покрытые клатрином.[6] Между тем, внутриклеточный белок, Dab1, имеет PI / PTB домен который взаимодействует с последовательностью NPxY, обнаруженной в цитоплазматическом хвосте VLDLR.[12] В результате Dab1 фосфорилируется по тирозину, а рилин разрушается. Наконец, фосфорилированный Dab1 активирует внутриклеточный сигнальный каскад, который направляет нейробласты в их правильное местоположение посредством изменения цитоскелет.[12][14] Многие особенности этого пути все еще исследуются. Пока не известно, фосфорилируется ли Dab1 в результате эндоцитоза рилина или задействован другой механизм. Помимо организации неокортекса, VLDLR также играет роль в миграции нейронов гиппокамп и Клетки Пуркинье из мозжечок. Тем не менее, большая часть информации об этом процессе до сих пор неизвестна.[6]
Сопутствующие расстройства
Мутации внутри VLDLR ген приводят к множеству расстройств разной степени тяжести. Эти расстройства обычно связаны с холестерин гомеостаз или нарушение порядка нейронов в головном мозге из-за нарушения катушка путь. Наиболее известные из этих заболеваний - тип I. лиссэнцефалия, Связанные с VLDR гипоплазия мозжечка, и атеросклероз. В отличие от заболеваний, VLDLR также считается возможным лекарством от некоторых расстройств. Внедрение ЛПОНП в печень может вылечить семейная гиперхолестеринемия (FH) у пациентов с дефектным LDLR или имеете дефектную иммунную систему, которая атакует этот белок. Поскольку VLDLR неиммуногенен, он не инициирует иммунный ответ, поэтому он может нормально функционировать при дефектной иммунной системе.[7] Кроме того, будучи тем апоЕ, главный лиганд VLDLR, является ведущим генетическим фактором риска развития Болезнь Альцгеймера, VLDLR могут играть роль в модуляции риска этого расстройства.[6] Было также показано, что VLDLR снижает вероятность преждевременных сердечных заболеваний и инсульта, поскольку VLDLR устраняется. липопротеин А (Lp (a)), основной наследственный фактор риска этих заболеваний.[7]
Лиссэнцефалия 1 типа
Тип I лиссэнцефалия, или агирия-пахигирия, представляет собой редкое нарушение развития, характеризующееся отсутствием извилины и борозды в мозгу. Эти серьезные пороки развития являются результатом неправильного миграция нейронов. При классической лизэнцефалии I типа миграция нейронов начинается, но не может продолжаться до конца. Этот процесс, вероятно, нарушен изменениями нескольких генов, включая VLDLR, DCX, ARX, TUBA1A, RELN и LIS1. Следовательно, тяжесть лизэнцефалии I типа зависит от типа мутации. Гомозиготная делеция, затрагивающая VLDLR гена приводит к низкой степени утолщения коры и отсутствию разреженной зоны. Клеточно-разреженная зона описывает область между внешним и внутренним кортикальными слоями арестованных нейронов.[15] Кроме того, лиссэнцефалия 1 типа тесно связана с гипоплазия мозжечка.
Гипоплазия мозжечка, связанная с ЛПОНП
Синдром неравновесия (DES) был впервые описан в 1970-х годах как непрогрессирующее неврологическое расстройство.[16] В исследовании 2005 года DES был переименован в Гипоплазия мозжечка, связанная с ЛПОНП (VLDLRCH) после того, как его причина была связана с нарушением VLDLR ген.[17] Не менее шести мутаций, влияющих на гомозиготный рецессивный аллель VLDLR были идентифицированы и обнаружено, что он вызывает VLDLRCH. Некоторые из этих мутаций были локализованы в определенных экзоны кодирующий ген. Одна из таких мутаций - это цитозин к тимин переход в паре оснований 1342 в экзоне 10, который вызывает замену в Arg 448 за сигнал завершения. Аналогичным образом, есть свидетельства перехода цитозина в тимин в паре оснований номер 769 в экзоне 5, который вызывает замену в Arg 257 для сигнала завершения. Третья известная мутация вызвана гомозиготной делецией 1 пары оснований в экзоне 17, которая вызывает сдвиг рамки и преждевременное прекращение О-связанный сахар домен.[18] Все подобные переделки в VLDLR ген предотвращает производство VLDLR и поэтому называется мутациями потери функции. Признанные симптомы VLDLRCH - умственная отсталость от умеренной до тяжелой степени, судороги, дизартрия, косоглазие и задержка передвижения. В некоторых случаях дети с VLDLRCH учатся ходить на очень позднем этапе развития после шести лет или никогда не учатся ходить самостоятельно. Частота этого расстройства неизвестна, поскольку ранняя диагностика VLDLRCH затруднена с использованием методов визуализации. Это связано с родительским кровное родство и встречаются в уединенных сообществах, таких как Гуттериты и инбредные семьи из Ирана и Турции.[19]
Атеросклероз
Атеросклероз отмечается чрезмерным накоплением холестерин к макрофаги, что привело к их превращению в пенные ячейки. Это накопление холестерина вызвано нарушением регуляции притока и оттока холестерина. Поскольку макрофаги не способны ограничивать приток холестерина, баланс полностью зависит от путей оттока. VLDLR экспрессируется макрофагами и функционирует при поглощении нативных липопротеины. Уникально, что VLDLR не реагирует на нагрузку холестерина, вероятно, из-за отсутствия механизмов обратной связи. Неспособность контролировать поглощение нативных липопротеинов делает VLDLR проатерогенным фактором.[20] Эта характеристика подтверждается результатами исследования 2005 г., в котором повторное введение VLDLR в VLDLR нокаутные мыши приводили к значительному увеличению развития атеросклеротических поражений.[20]
Смотрите также
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000147852 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск ансамбля 89: ENSMUSG00000024924 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Nimpf J, Schneider WJ (декабрь 2000 г.). «От транспорта холестерина к передаче сигнала: рецептор липопротеинов низкой плотности, рецептор липопротеинов очень низкой плотности и рецептор-2 аполипопротеина E». Биохим. Биофиз. Acta. 1529 (1–3): 287–98. Дои:10.1016 / S1388-1981 (00) 00155-4. PMID 11111096.
- ^ а б c d е ж грамм час я j k л м п о Редди СС, Коннор Т.Э., Уибер Э.Дж., Ребек В. (2011). «Сходства и различия в структуре, экспрессии и функциях VLDLR и ApoER2». Мол нейродегенератор. 6: 30. Дои:10.1186/1750-1326-6-30. ЧВК 3113299. PMID 21554715.
- ^ а б c d е ж грамм час я Такахаши С., Сакаи Дж., Фуджино Т., Хаттори Х., Зенимару Ю., Сузуки Дж., Миямори И., Ямамото Т. Т. (2004). «Рецептор липопротеинов очень низкой плотности (ЛПОНП): характеристика и функции рецептора периферических липопротеинов». J. Atheroscler. Тромб. 11 (4): 200–8. Дои:10.5551 / jat.11.200. PMID 15356379.
- ^ а б c d е ж грамм Go GW, Mani A (март 2012 г.). «Семейство рецепторов липопротеинов низкой плотности (ЛПНП) регулирует гомеостаз холестерина». Йель Дж Биол Мед. 85 (1): 19–28. ЧВК 3313535. PMID 22461740.
- ^ Тиссир Ф., Гоффине А.М. (июнь 2003 г.). «Рилин и развитие мозга». Nat. Преподобный Neurosci. 4 (6): 496–505. Дои:10.1038 / nrn1113. PMID 12778121. S2CID 12039624.
- ^ а б Nimpf J, Schneider WJ (декабрь 1998 г.). «Рецептор VLDL: родственник рецептора LDL с восемью повторами связывания лиганда, LR8». Атеросклероз. 141 (2): 191–202. Дои:10.1016 / с0021-9150 (98) 00172-5. PMID 9862168.
- ^ а б c d е GUPEA: механизмы и последствия накопления липидов в клетках - роль рецептора липопротеинов очень низкой плотности (ЛПОНП). 2011-12-02. HDL:2077/27815. ISBN 9789162883560.
- ^ а б c Райс Д.С., Курран Т. (2001). «Роль сигнального пути рилина в развитии центральной нервной системы». Анну. Преподобный Neurosci. 24: 1005–39. Дои:10.1146 / annurev.neuro.24.1.1005. PMID 11520926. S2CID 17258257.
- ^ а б Валиенте М., Марин О. (февраль 2010 г.). «Механизмы миграции нейронов в развитии и болезни». Curr. Мнение. Нейробиол. 20 (1): 68–78. Дои:10.1016 / j.conb.2009.12.003. PMID 20053546. S2CID 18658808.
- ^ Биелас С., Хиггинботам Х, Коидзуми Х, Танака Т., Глисон Дж. Г. (2004). «Мутанты миграции корковых нейронов предполагают отдельные, но пересекающиеся пути». Анну. Rev. Cell Dev. Биол. 20: 593–618. Дои:10.1146 / annurev.cellbio.20.082503.103047. PMID 15473853.
- ^ Спалис А., Паризи П., Никита Ф., Пиццарди Дж., Дель Бальцо Ф., Яннетти П. (март 2009 г.). «Нарушения миграции нейронов: клинические, нейрорадиологические и генетические аспекты». Acta Paediatr. 98 (3): 421–33. Дои:10.1111 / j.1651-2227.2008.01160.x. PMID 19120042. S2CID 21620197.
- ^ Мохеб Л.А., Цшах А., Гаршасби М., Кахризи К., Дарвиш Х., Хешмати Й., Корди А., Наджмабади Х., Роперс Х. Х., Кусс А. В. (февраль 2008 г.). «Идентификация нонсенс-мутации в гене рецептора липопротеинов очень низкой плотности (VLDLR) в иранской семье с синдромом дисбаланса». Евро. J. Hum. Genet. 16 (2): 270–3. Дои:10.1038 / sj.ejhg.5201967. PMID 18043714.
- ^ Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR, Parboosingh JS (сентябрь 2005 г.). «Гомозиготная делеция гена рецептора липопротеинов очень низкой плотности вызывает аутосомно-рецессивную гипоплазию мозжечка с упрощением церебральной гирали». Являюсь. J. Hum. Genet. 77 (3): 477–83. Дои:10.1086/444400. ЧВК 1226212. PMID 16080122.
- ^ Онлайн-менделевское наследование в человеке (OMIM): Гипоплазия мозжечка, ассоциированная с VLDLR; VLDLRCH - 224050
- ^ Бойкот К. М., Парбоусинг Дж. С. (2008). «Гипоплазия мозжечка, связанная с VLDLR». В Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (ред.). GeneReviews [Интернет]. PMID 20301729.
- ^ а б Пеннингс М., Меурс I, Йе Д., Аут Р., Хёкстра М., Ван Беркель Т.Дж., Ван Эк М (октябрь 2006 г.). «Регуляция гомеостаза холестерина в макрофагах и последствия развития атеросклеротического поражения». FEBS Lett. 580 (23): 5588–96. Дои:10.1016 / j.febslet.2006.08.022. PMID 16935283. S2CID 42158329.
дальнейшее чтение
- Ока К., Ишимура-Ока К., Чу М.Дж., Салливан М., Крушкал Дж., Ли У.Х., Чан Л. (сентябрь 1994 г.). «Клонирование кДНК рецептора липопротеинов очень низкой плотности (VLDLR) мыши, тканеспецифическая экспрессия и эволюционная связь с рецептором липопротеинов низкой плотности». Евро. J. Biochem. 224 (3): 975–82. Дои:10.1111 / j.1432-1033.1994.00975.x. PMID 7925422.
- Ананьева Н.М., Макогоненко Ю.М., Куявская Д.В., Руис Дж., Лимбург В., Мейер А.Б., Хренов А.В., Шима М., Стрикленд Д.К., Саенко Е.Л. (март 2008 г.). «Сайты связывания для рецептора липопротеинов очень низкой плотности и белка, родственного рецептору липопротеинов низкой плотности, являются общими для фактора свертывания крови VIII». Сгусток крови. Фибринолиз. 19 (2): 166–77. Дои:10.1097 / MBC.0b013e3282f5457b. PMID 18277139. S2CID 10380641.
- Ананьева Н.М., Макогоненко Ю.М., Сарафанов А.Г., Печик И.В., Горлатова Н., Радтке К.П., Шима М., Саенко Е.Л. (сентябрь 2008 г.). «Взаимодействие фактора свертывания крови VIII с членами семейства рецепторов липопротеинов низкой плотности следует по общему механизму и включает консенсусные остатки в участке связывания А2 484-509». Сгусток крови. Фибринолиз. 19 (6): 543–55. Дои:10.1097 / MBC.0b013e3283068859. PMID 18685438. S2CID 31127950.
- Льорка Дж., Родригес-Родригес Э., Диерсен-Сотос Т., Дельгадо-Родригес М., Берчиано Дж., Комбаррос О. (январь 2008 г.).«Мета-анализ генетической изменчивости в метаболических путях продукции, агрегации и деградации бета-амилоида и риска болезни Альцгеймера». Acta Neurol. Сканд. 117 (1): 070914011339003––. Дои:10.1111 / j.1600-0404.2007.00899.x. PMID 17854420. S2CID 25781860.
- Озчелик Т., Акарсу Н., Уз Э, Чаглаян С., Гульсунер С., Онат О. Е., Тан М., Тан У (март 2008 г.). «Мутации в рецепторе липопротеинов очень низкой плотности VLDLR вызывают гипоплазию мозжечка и передвижение на четвероногих лапах у людей». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 105 (11): 4232–6. Дои:10.1073 / pnas.0710010105. ЧВК 2393756. PMID 18326629.
- Türkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, Mundlos S (сентябрь 2008 г.). «Гипоплазия мозжечка с движением на четвероногих ногах, вызванная мутациями в гене рецептора липопротеинов очень низкой плотности». Евро. J. Hum. Genet. 16 (9): 1070–4. Дои:10.1038 / ejhg.2008.73. PMID 18364738.
- Оганесян А., Армстронг Л.С., Мильорини М.М., Стрикленд Д.К., Борнштейн П. (февраль 2008 г.). «Тромбоспондины используют рецептор VLDL и неапоптотический путь для подавления деления клеток в эндотелиальных клетках микрососудов». Мол. Биол. Клетка. 19 (2): 563–71. Дои:10.1091 / mbc.E07-07-0649. ЧВК 2230579. PMID 18032585.
- Wruss J, Rünzler D, Steiger C, Chiba P, Köhler G, Blaas D (май 2007 г.). «Присоединение рецепторов VLDL к икосаэдрическому вирусу вдоль 5-кратной оси симметрии: множественные способы связывания, подтвержденные флуоресцентной корреляционной спектроскопией». Биохимия. 46 (21): 6331–9. Дои:10.1021 / bi700262w. PMID 17472347.
- Судзуки К., Накамура К., Ивата И., Секин Ю., Кавай М., Сугихара Г., Цучия К.Дж., Суда С., Мацудзаки Н., Такей Н., Хашимото К., Мори Н. (январь 2008 г.). «Снижение экспрессии рецептора рилина VLDLR в периферических лимфоцитах у больных шизофренией, не принимавших лекарственные препараты». Schizophr. Res. 98 (1–3): 148–56. Дои:10.1016 / j.schres.2007.09.029. PMID 17936586. S2CID 45594329.
- Фрэнсис П.Дж., Хамон С.К., Отт Дж., Велебер Р.Г., Кляйн М.Л. (май 2009 г.). «Полиморфизмы в C2, CFB и C3 связаны с прогрессированием к пожилой возрастной дегенерации желтого пятна, связанной с потерей зрения». J. Med. Genet. 46 (5): 300–7. Дои:10.1136 / jmg.2008.062737. PMID 19015224. S2CID 22940548.
- Чжан Г., Ассади А.Х., Макнил Р.С., Бефферт Ю., Виншоу-Борис А., Герц Дж., Кларк Г.Д., Д'Арканджело Г. (2007). «Комплекс Pafah1b взаимодействует с рецептором рилина VLDLR». PLOS ONE. 2 (2): e252. Дои:10.1371 / journal.pone.0000252. ЧВК 1800349. PMID 17330141.
- Пуарье С., Майер Дж., Бенджаннет С., Бержерон Э, Марцинкевич Дж., Нассури Н., Майер Х., Нимпф Дж., Прат А., Сейда Н. Г. (январь 2008 г.). «Пропротеинконвертаза PCSK9 вызывает деградацию рецептора липопротеинов низкой плотности (LDLR) и его ближайших членов семейства VLDLR и ApoER2». J. Biol. Chem. 283 (4): 2363–72. Дои:10.1074 / jbc.M708098200. PMID 18039658.
- Crawford DC, Nord AS, Badzioch MD, Ranchalis J, McKinstry LA, Ahearn M, Bertucci C, Shephard C, Wong M, Rieder MJ, Schellenberg GD, Nickerson DA, Heagerty PJ, Wijsman EM, Jarvik GP (март 2008 г.). «Обычный полиморфизм VLDLR взаимодействует с генотипом APOE в прогнозировании риска заболевания сонной артерии». J. Lipid Res. 49 (3): 588–96. Дои:10.1194 / мл. M700409-JLR200. PMID 18056683.
- Ямада Y, Андо Ф, Симоката Х (июль 2005 г.). «Ассоциация полиморфизмов CYP17A1, MTP и VLDLR с минеральной плотностью костей у японских женщин и мужчин, проживающих в общинах». Геномика. 86 (1): 76–85. Дои:10.1016 / j.ygeno.2005.03.005. PMID 15953542.
- Чен Й, Ху Й, Лу К., Фланнери Дж. Дж., Ма Дж. Х (ноябрь 2007 г.). «Рецептор липопротеинов очень низкой плотности, негативный регулятор пути передачи сигналов wnt и хориоидальной неоваскуляризации». J. Biol. Chem. 282 (47): 34420–8. Дои:10.1074 / jbc.M611289200. PMID 17890782.
- Haines JL, Schnetz-Boutaud N, Schmidt S, Scott WK, Agarwal A, Postel EA, Olson L, Kenealy SJ, Hauser M, Gilbert JR, Pericak-Vance MA (январь 2006 г.). «Функциональные гены-кандидаты при возрастной дегенерации желтого пятна: значимая связь с VEGF, VLDLR и LRP6». Вкладывать деньги. Офтальмол. Vis. Наука. 47 (1): 329–35. Дои:10.1167 / iovs.05-0116. PMID 16384981.
- Сакаи К., Тибель О., Юнгберг М.К., Салливан М., Ли Х.Дж., Терашима Т., Ли Р., Кобаяши К., Лу ХК, Чан Л., Ока К. (июнь 2009 г.). «Вариант нейрональных VLDLR, лишенный третьего повтора типа комплемента, демонстрирует высокую способность связывания липопротеинов, содержащих ароЕ». Brain Res. 1276: 11–21. Дои:10.1016 / j.brainres.2009.04.030. ЧВК 2733343. PMID 19393635.
- Мозер Р., Снайерс Л., Врусс Дж., Ангуло Дж., Петерс Х., Петерс Т., Блаас Д. (август 2005 г.). «Нейтрализация вируса простуды конкатемерами третьего модуля связывания лиганда ЛПОНП-рецептора сильно зависит от количества модулей». Вирусология. 338 (2): 259–69. Дои:10.1016 / j.virol.2005.05.016. PMID 15950998.
внешняя ссылка
- GeneReviews / NCBI / NIH / UW статья о VLDLR-ассоциированной гипоплазии мозжечка или синдроме дисбаланса - VLDLR
- Обзор всей структурной информации, доступной в PDB за UniProt: P98155 (Рецептор липопротеинов очень низкой плотности) на PDBe-KB.