РНК-Seq - RNA-Seq

РНК-Seq (названный аббревиатурой от «секвенирования РНК») - это особая технология, основанная на последовательность действий техника, которая использует секвенирование следующего поколения (NGS), чтобы выявить наличие и количество РНК в биологическом образце в данный момент, анализируя непрерывно изменяющиеся клеточные транскриптом.[2][3]
В частности, RNA-Seq облегчает возможность просмотра транскрипты с альтернативным сплайсингом генов, посттранскрипционные модификации, слияние генов, мутации /SNP и изменения в экспрессии генов с течением времени или различия в экспрессии генов в разных группах или в разных обработках.[4] Помимо транскриптов мРНК, RNA-Seq может анализировать различные популяции РНК, включая общую РНК, малую РНК, такую как miRNA, тРНК, и рибосомное профилирование.[5] RNA-Seq также может использоваться для определения экзон /интрон границы и проверить или изменить ранее аннотированный 5' и 3' границы генов. Последние достижения в области RNA-Seq включают: секвенирование одной клетки и секвенирование фиксированной ткани на месте.[6]
До RNA-Seq исследования экспрессии генов проводились с помощью гибридизации. микрочипы. Проблемы с микрочипами включают артефакты перекрестной гибридизации, плохую количественную оценку низко и высоко экспрессируемых генов и необходимость знать последовательность. априори.[7] Из-за этих технических проблем транскриптомика перешли на методы секвенирования. Они произошли от Секвенирование по Сэнгеру из Выраженный тег последовательности библиотеки, к методам на основе химических меток (например, серийный анализ экспрессии генов ), и, наконец, к современной технологии, секвенирование следующего поколения из кДНК (особенно РНК-Seq).
Методы
Подготовка библиотеки

Общие шаги по подготовке комплементарная ДНК (cDNA) библиотеки для секвенирования описаны ниже, но часто различаются между платформами.[8][3][9]
- Выделение РНК: РНК изолирована из ткани и смешанный с дезоксирибонуклеаза (ДНКаза). ДНКаза уменьшает количество геномной ДНК. Степень деградации РНК проверяется с помощью гель и капиллярный электрофорез и используется для присвоения Число целостности РНК к образцу. Это качество РНК и общее количество исходной РНК учитываются на последующих этапах подготовки библиотеки, секвенирования и анализа.
- Выбор / истощение РНК: Для анализа представляющих интерес сигналов выделенную РНК можно оставить как есть, лишенную рибосомная РНК (рРНК), фильтруется на РНК с 3 'полиаденилированный (поли (А)) хвосты включать только мРНК, и / или отфильтрованы для РНК, которая связывает определенные последовательности (Методы отбора и истощения РНК Таблица ниже). У эукариот РНК с 3'-поли (A) хвостами представляет собой зрелые процессированные кодирующие последовательности. Селекция поли (А) выполняется путем смешивания эукариотической РНК с поли (Т) олигомерами, ковалентно прикрепленными к субстрату, обычно магнитным шарикам.[10][11] Выбор поли (A) игнорирует некодирующую РНК и вводит 3 'смещение,[12] чего можно избежать с помощью стратегии истощения рибосом. РРНК удаляется, потому что она составляет более 90% РНК в клетке, и если ее сохранить, то другие данные в транскриптоме будут скрыты.
- синтез кДНК: РНК обратная расшифровка в кДНК, потому что ДНК более стабильна и позволяет амплификацию (которая использует ДНК-полимеразы ) и использовать более совершенную технологию секвенирования ДНК. Амплификация после обратной транскрипции приводит к потере замкнутость, чего можно избежать с помощью химического мечения или секвенирования одной молекулы. Фрагментация и выбор размера выполняются для очистки последовательностей, которые имеют подходящую длину для секвенатора. РНК, кДНК или обе фрагментированы ферментами, обработка ультразвуком, или небулайзеры. Фрагментация РНК снижает 5'-смещение произвольно праймированной обратной транскрипции и влияние грунтовка участок связывания,[11] с другой стороны, 5 'и 3' концы превращаются в ДНК менее эффективно. За фрагментацией следует выбор размера, при котором либо удаляются небольшие последовательности, либо выбирается узкий диапазон длин последовательностей. Поскольку малые РНК, такие как миРНК теряются, они анализируются независимо. КДНК для каждого эксперимента может быть проиндексирована штрих-кодом гексамера или октамера, так что эти эксперименты могут быть объединены в одну дорожку для мультиплексного секвенирования.
| Стратегия | Тип РНК | Содержание рибосомальной РНК | Необработанное содержание РНК | Содержание геномной ДНК | Метод изоляции |
|---|---|---|---|---|---|
| Общая РНК | Все | Высоко | Высоко | Высоко | Никто |
| Выбор PolyA | Кодирование | Низкий | Низкий | Низкий | Гибридизация с поли (dT) олигомеры |
| истощение рРНК | Кодирование, некодирование | Низкий | Высоко | Высоко | Удаление олигомеров, комплементарных рРНК |
| Захват РНК | Целевые | Низкий | Умеренный | Низкий | Гибридизация с зондами, комплементарными желаемым транскриптам |
Секвенирование малой / некодирующей РНК
При секвенировании РНК, отличной от мРНК, подготовка библиотеки изменяется. Клеточная РНК выбирается на основе желаемого диапазона размеров. Для малых мишеней РНК, таких как miRNA, РНК выделяется путем отбора по размеру. Это может быть выполнено с помощью геля для исключения размера, с помощью магнитных шариков для выбора размера или с помощью коммерчески разработанного набора. После выделения линкеры добавляют к 3'- и 5'-концам, а затем очищают. Последний шаг - кДНК генерация посредством обратной транскрипции.
Прямое секвенирование РНК

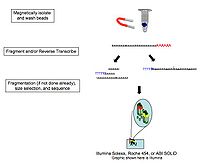
Поскольку преобразование РНК в кДНК было показано, что лигирование, амплификация и другие манипуляции с образцами вносят систематические ошибки и артефакты, которые могут мешать как правильной характеристике, так и количественной оценке транскриптов,[13] прямое секвенирование одной молекулы РНК было исследовано компаниями, включая Геликос (банкрот), Oxford Nanopore Technologies,[14] и другие. Эта технология обеспечивает прямую последовательность молекул РНК массово-параллельным образом.
Секвенирование одноклеточной РНК (scRNA-Seq)
Стандартные методы, такие как микрочипы и стандартный объемный анализ РНК-Seq анализирует экспрессию РНК из больших популяций клеток. В смешанных популяциях клеток эти измерения могут скрыть важные различия между отдельными клетками в этих популяциях.[15][16]
Секвенирование одноклеточной РНК (scRNA-Seq) обеспечивает профили выражения отдельных ячеек. Хотя невозможно получить полную информацию о каждой РНК, экспрессируемой каждой клеткой, из-за небольшого количества доступного материала, паттерны экспрессии генов могут быть идентифицированы через ген кластерный анализ. Это может раскрыть существование в популяции клеток редких типов клеток, которые, возможно, никогда раньше не наблюдались. Например, редкие специализированные клетки в легких, называемые легочные ионоциты которые выражают Регулятор трансмембранной проводимости при кистозном фиброзе были идентифицированы в 2018 году двумя группами, выполняющими scRNA-Seq на эпителии дыхательных путей легких.[17][18]
Экспериментальные процедуры

Текущие протоколы scRNA-Seq включают следующие этапы: выделение отдельной клетки и РНК, обратная транскрипция (RT), амплификация, создание библиотеки и секвенирование. Ранние методы разделяли отдельные клетки на отдельные лунки; более современные методы инкапсулируют отдельные клетки в капельки в микрофлюидном устройстве, где происходит реакция обратной транскрипции, превращающая РНК в кДНК. Каждая капля несет «штрих-код» ДНК, который однозначно маркирует кДНК, полученные из одной клетки. После завершения обратной транскрипции кДНК из многих клеток могут быть смешаны вместе для секвенирования; транскрипты из конкретной клетки идентифицируются уникальным штрих-кодом.[19][20]
Проблемы для scRNA-Seq включают сохранение исходного относительного количества мРНК в клетке и идентификацию редких транскриптов.[21] Этап обратной транскрипции имеет решающее значение, поскольку эффективность реакции RT определяет, какая часть популяции РНК клетки будет в конечном итоге проанализирована секвенатором. Процессивность обратных транскриптаз и используемые стратегии прайминга могут влиять на продукцию полноразмерной кДНК и создание библиотек, смещенных в сторону 3 ’или 5’ конца генов.
На этапе амплификации либо ПЦР, либо in vitro транскрипция (IVT) в настоящее время используется для амплификации кДНК. Одним из преимуществ методов на основе ПЦР является возможность генерировать полноразмерную кДНК. Однако различная эффективность ПЦР для конкретных последовательностей (например, содержимого GC и структуры snapback) также может быть экспоненциально усилена, создавая библиотеки с неравномерным покрытием. С другой стороны, хотя библиотеки, созданные с помощью IVT, могут избежать смещения последовательности, вызванного ПЦР, определенные последовательности могут транскрибироваться неэффективно, что вызывает выпадение последовательности или генерирование неполных последовательностей.[22][15]Было опубликовано несколько протоколов scRNA-Seq: Tang et al.,[23]STRT,[24]SMART-seq,[25]CEL-seq,[26]RAGE-seq,[27], Кварц-сек.[28]и C1-CAGE.[29] Эти протоколы различаются с точки зрения стратегий обратной транскрипции, синтеза и амплификации кДНК, а также возможностью размещения штрих-кодов, специфичных для последовательности (т.е. UMI ) или способность обрабатывать объединенные образцы.[30]
В 2017 году были внедрены два подхода для одновременного измерения экспрессии мРНК и белка в отдельных клетках с помощью меченных олигонуклеотидами антител, известных как REAP-seq,[31] и CITE-seq.[32]
Приложения
scRNA-Seq широко используется в биологических дисциплинах, включая разработку, Неврология,[33] Онкология,[34][35][36] Аутоиммунное заболевание,[37] и Инфекционное заболевание.[38]
scRNA-Seq предоставил значительное понимание развития эмбрионов и организмов, включая червя Caenorhabditis elegans,[39] и регенеративная планария Schmidtea mediterranea.[40][41] Первыми позвоночными животными, которые были нанесены на карту таким образом, были Данио[42][43] и Xenopus laevis.[44] В каждом случае изучались несколько стадий эмбриона, что позволяло картировать весь процесс развития на клеточной основе.[8] Наука признал эти достижения 2018 г. Прорыв года.[45]
Экспериментальные соображения
Разнообразие параметры учитываются при разработке и проведении экспериментов по RNA-Seq:
- Тканевая специфичность: Экспрессия генов варьируется в пределах и между тканями, и RNA-Seq измеряет это сочетание типов клеток. Это может затруднить выделение интересующего биологического механизма. Секвенирование одной клетки можно использовать для изучения каждой ячейки в отдельности, что устраняет эту проблему.
- Временная зависимость: Экспрессия генов меняется со временем, и RNA-Seq делает только снимок. Можно проводить эксперименты с течением времени, чтобы наблюдать изменения в транскриптоме.
- Покрытие (также известное как глубина): РНК содержит те же мутации, которые наблюдаются в ДНК, и для обнаружения требуется более глубокий охват. При достаточно высоком покрытии RNA-Seq можно использовать для оценки экспрессии каждого аллеля. Это может дать представление о таких явлениях, как печать или цис-регуляторные эффекты. Глубина секвенирования, необходимая для конкретных приложений, может быть экстраполирована из пилотного эксперимента.[46]
- Артефакты генерации данных (также известные как технические отклонения): Реагенты (например, набор для подготовки библиотеки), задействованный персонал и тип секвенатора (например, Иллюмина, Тихоокеанские биологические науки ) может привести к техническим артефактам, которые могут быть неверно интерпретированы как значимые результаты. Как и любой научный эксперимент, разумно проводить RNA-Seq в хорошо контролируемой обстановке. Если это невозможно или исследование является метаанализ, другое решение - обнаруживать технические артефакты путем вывода скрытые переменные (обычно Анализ главных компонентов или факторный анализ ) с последующей поправкой на эти переменные.[47]
- Управление данными: Единичный эксперимент RNA-Seq на людях обычно проводится в порядке 1 Гб.[48] Такой большой объем данных может создавать проблемы с хранением. Одно из решений сжатие данные с использованием многоцелевых вычислительных схем (например, gzip ) или специфичные для геномики схемы. Последние могут быть основаны на эталонных последовательностях или de novo. Другое решение - провести эксперименты с микрочипами, которых может быть достаточно для работы, основанной на гипотезах, или исследований репликации (в отличие от исследовательских исследований).
Анализ
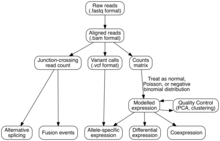
Сборка транскриптома
Два метода используются для присвоения считывания необработанной последовательности геномным признакам (т. Е. Сборки транскриптома):
- De novo: Такой подход не требует эталонный геном для реконструкции транскриптома и обычно используется, если геном неизвестен, неполный или существенно изменен по сравнению с эталоном.[49] Проблемы при использовании коротких чтений для сборки de novo включают: 1) определение того, какие чтения следует объединить в непрерывные последовательности (контиги ), 2) устойчивость к ошибкам секвенирования и другим артефактам, и 3) вычислительная эффективность. Основной алгоритм, используемый для сборки de novo, перешел от графов перекрытия, которые идентифицируют все попарные перекрытия между чтениями, к графы де Брейна, который разбивает чтение на последовательности длины k и сворачивает все k-мерки в хеш-таблицу.[50] Графики перекрытия использовались при секвенировании по Сэнгеру, но они плохо масштабируются до миллионов считываний, созданных с помощью RNA-Seq. Примеры ассемблеров, использующих графы де Брейна: Бархат,[51] Троица,[49] Оазисы,[52] и Бриджер.[53] Парное секвенирование конца и длинное чтение одного и того же образца может уменьшить недостатки в коротком секвенировании чтения, выступая в качестве шаблона или скелета. Метрики для оценки качества сборки de novo включают среднюю длину контигов, количество контигов и N50.[54]
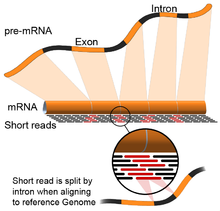
- Направляемый геном: Этот подход основан на тех же методах, что и для выравнивания ДНК, с дополнительной сложностью выравнивания считываний, которые охватывают прерывистые части эталонного генома.[55] Эти прерывистые чтения являются результатом секвенирования склеенных транскриптов (см. Рисунок). Как правило, алгоритмы выравнивания состоят из двух этапов: 1) выравнивание коротких участков считываемых данных (т. Е. Засеивание генома) и 2) использование динамическое программирование чтобы найти оптимальное выравнивание, иногда в сочетании с известными аннотациями. Программные инструменты, использующие геномное выравнивание, включают Bowtie,[56] TopHat (который основан на результатах BowTie для выравнивания стыков),[57][58] Subread,[59] ЗВЕЗДА,[55] HISAT2,[60] Парусник,[61] Каллисто,[62] и GMAP.[63] Качество управляемой геномом сборки можно измерить с помощью 1) показателей сборки de novo (например, N50) и 2) сравнений с известным транскриптом, сплайсинговыми соединениями, геномными и белковыми последовательностями с использованием точность, отзыв, или их комбинация (например, оценка F1).[54] К тому же, in silico оценка может быть выполнена с использованием имитационных чтений.[64][65]
Замечание по качеству сборки: Текущий консенсус заключается в том, что 1) качество сборки может варьироваться в зависимости от того, какая метрика используется, 2) сборки, получившие хорошие оценки у одного вида, не обязательно хорошо работают у другого вида, и 3) сочетание различных подходов может быть наиболее надежным.[66][67]
Количественная оценка экспрессии генов
Экспрессия оценивается количественно для изучения клеточных изменений в ответ на внешние раздражители, различий между здоровыми и здоровыми. больной состояния и другие вопросы исследования. Экспрессия генов часто используется в качестве показателя обилия белка, но они часто не эквивалентны из-за посттранскрипционных событий, таких как РНК-интерференция и бессмысленный распад.[68]
Выражение количественно оценивается путем подсчета числа считываний, сопоставленных с каждым локусом в сборка транскриптома шаг. Экспрессию экзонов или генов можно количественно оценить с помощью контигов или аннотаций эталонных транскриптов.[8] Эти наблюдаемые подсчеты считывания RNA-Seq были тщательно проверены на соответствие более старым технологиям, включая экспрессионные микрочипы и КПЦР.[46][69] Примеры инструментов для количественной оценки подсчетов: HTSeq,[70] FeatureCounts,[71] Rcount,[72] maxcounts,[73] FIXSEQ,[74] и Cuffquant. Счетчики чтения затем преобразуются в соответствующие показатели для проверки гипотез, регрессии и других анализов. Параметры для этого преобразования:
- Глубина секвенирования / охват: Хотя глубина предварительно указывается при проведении нескольких экспериментов с RNA-Seq, она все равно будет сильно различаться между экспериментами.[75] Таким образом, общее количество считываний, сгенерированных в одном эксперименте, обычно нормализуется путем преобразования счетчиков во фрагменты, чтения или счета на миллион отображенных считываний (FPM, RPM или CPM). Глубину секвенирования иногда называют размер библиотеки - количество промежуточных молекул кДНК в эксперименте.
- Длина гена: Более длинные гены будут иметь больше фрагментов / считываний / отсчетов, чем более короткие гены, если экспрессия транскриптов одинакова. Это регулируется путем деления FPM на длину гена, в результате чего получается количество метрических фрагментов на килобазу транскрипта на миллион отображенных считываний (FPKM).[76] При просмотре групп генов в образцах FPKM преобразуется в количество транскриптов на миллион (TPM) путем деления каждого FPKM на сумму FPKM в образце.[77][78][79]
- Общий выход образца РНК: Поскольку из каждого образца извлекается одинаковое количество РНК, образцы с большим количеством общей РНК будут иметь меньше РНК на ген. Эти гены, по-видимому, имеют пониженную экспрессию, что приводит к ложноположительным результатам последующих анализов.[75] Стратегии нормализации, включая квантиль, DESeq2, TMM и Median Ratio, пытаются учесть это различие путем сравнения набора генов, экспрессируемых недифференциально, между выборками и соответствующего масштабирования.[80]
- Дисперсия для выражения каждого гена: моделируется с учетом ошибка выборки (важно для генов с низким числом считываний), увеличить мощность и уменьшить количество ложных срабатываний. Дисперсию можно оценить как нормальный, Пуассон, или же отрицательный бином распространение[81][82][83] и часто разлагается на техническую и биологическую вариации.
Абсолютная количественная оценка
Абсолютная количественная оценка экспрессии генов невозможна в большинстве экспериментов с RNA-Seq, которые определяют количественную экспрессию относительно всех транскриптов. Возможно, выполнив RNA-Seq со вставками, образцы РНК в известных концентрациях. После секвенирования счетчики считываний всплесков последовательностей используются для определения взаимосвязи между счетчиками считываний каждого гена и абсолютными количествами биологических фрагментов.[11][84] В одном примере этот метод использовался в Xenopus tropicalis эмбрионов для определения кинетики транскрипции.[85]
Дифференциальное выражение
Самое простое, но часто наиболее эффективное использование RNA-Seq - это обнаружение различий в экспрессии генов между двумя или более состояниями (например, лечится vs не лечится); этот процесс называется дифференциальным выражением. Выходы часто называют дифференциально экспрессируемыми генами (ДЭГ), и эти гены могут регулироваться либо с повышением, либо с понижением (т.е., выше или ниже в интересующем состоянии). Есть много инструменты, которые выполняют дифференциальное выражение. Большинство из них запущено р, Python, или Unix командная строка. Обычно используемые инструменты включают DESeq,[82] крайR,[83] и вуом + лимма,[81][86] все это доступно через R /Биокондуктор.[87][88] Вот общие соображения при выполнении дифференциального выражения:
- Входы: Входы дифференциальной экспрессии включают (1) матрицу экспрессии RNA-Seq (M генов x N образцов) и (2) a матрица дизайна содержащие экспериментальные условия для N образцов. Простейшая матрица плана содержит один столбец, соответствующий меткам проверяемого условия. Другие ковариаты (также называемые факторами, функциями, метками или параметрами) могут включать пакетные эффекты, известные артефакты и любые метаданные, которые могут мешать или опосредовать экспрессию генов. В дополнение к известным ковариатам, неизвестные ковариаты также могут быть оценены с помощью неконтролируемое машинное обучение подходы, включая главный компонент, суррогатная переменная,[89] и PEER[47] анализы. Анализ скрытых переменных часто используется для данных RNA-Seq тканей человека, которые обычно содержат дополнительные артефакты, не зафиксированные в метаданных (например, время ишемии, источники из нескольких учреждений, основные клинические признаки, сбор данных за многие годы с большим количеством сотрудников).
- Методы: Большинство инструментов используют регресс или непараметрическая статистика для идентификации дифференциально экспрессируемых генов, которые основаны либо на подсчете (DESeq2, limma, edgeR), либо на сборке (с помощью количественной оценки без выравнивания, сыщика,[90] Cuffdiff,[91] Бальное платье[92]).[93] После регрессии большинство инструментов используют либо уровень семейных ошибок (FWER) или коэффициент ложного обнаружения (FDR) корректировки p-значения для учета несколько гипотез (в исследованиях на людях ~ 20 000 генов, кодирующих белок, или ~ 50 000 биотипов).
- Выходы: Типичный вывод состоит из строк, соответствующих количеству генов, и не менее трех столбцов, журнал каждого гена сложить изменение (лог-преобразование отношения в выражении между условиями, мера размер эффекта ), p-значение, и p-значение с поправкой на множественные сравнения. Гены считаются биологически значимыми, если они проходят пороговые значения для величины эффекта (изменение логарифмической кратности) и Статистическая значимость. В идеале эти границы должны быть указаны априори, но природа экспериментов с RNA-Seq часто носит исследовательский характер, поэтому сложно заранее предсказать размеры эффекта и соответствующие пороговые значения.
- Подводные камни: Смысл использования этих сложных методов состоит в том, чтобы избежать множества ловушек, которые могут привести к статистические ошибки и вводящие в заблуждение толкования. К подводным камням относятся увеличение числа ложноположительных результатов (из-за множественных сравнений), артефакты подготовки образцов, неоднородность образцов (например, смешанный генетический фон), высококоррелированные образцы, неучтенные многоуровневые экспериментальные конструкции и бедный экспериментальная конструкция. Одна заметная ошибка - просмотр результатов в Microsoft Excel без использования функции импорта, чтобы имена генов оставались текстовыми.[94] Хотя это удобно, Excel автоматически преобразует названия некоторых генов (1 СЕНТЯБРЯ, DEC1, 2 МАРТА ) в даты или числа с плавающей запятой.
- Выбор инструментов и сравнительный анализ: Существует множество попыток сравнить результаты этих инструментов, при этом DESeq2 имеет тенденцию умеренно превосходить другие методы.[95][96][97][98][99][93][100] Как и другие методы, сравнительный анализ состоит из сравнения результатов инструментов друг с другом и известных золотые стандарты.
Последующий анализ списка дифференциально экспрессируемых генов бывает двух видов: подтверждение наблюдений и биологические выводы. Из-за ловушек дифференциальной экспрессии и RNA-Seq важные наблюдения воспроизводятся (1) ортогональным методом в тех же образцах (например, ПЦР в реальном времени ) или (2) другой, иногда предварительно зарегистрированный, поэкспериментируйте в новой когорте. Последнее помогает обеспечить обобщаемость и, как правило, может сопровождаться метаанализом всех объединенных когорт. Наиболее распространенный метод получения биологического понимания результатов более высокого уровня - это анализ обогащения набора генов, хотя иногда используются подходы с генами-кандидатами. Обогащение набора генов определяет, является ли перекрытие между двумя наборами генов статистически значимым, в этом случае перекрытие между дифференциально экспрессируемыми генами и наборами генов из известных путей / баз данных (например, Генная онтология, КЕГГ, Онтология человеческого фенотипа ) или из дополнительных анализов тех же данных (например, сетей коэкспрессии). Общие инструменты для обогащения набора генов включают веб-интерфейсы (например, ENRICHR, g: profiler) и программные пакеты. При оценке результатов обогащения одна эвристика состоит в том, чтобы сначала искать обогащение известной биологии в качестве проверки работоспособности, а затем расширять область поиска для поиска новой биологии.
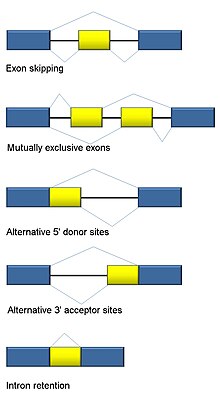
Альтернативная сварка
Сплайсинг РНК является неотъемлемой частью эукариот и вносит значительный вклад в регуляцию и разнообразие белков, присутствуя в> 90% генов человека.[101] Есть несколько альтернативные режимы сварки: пропуск экзонов (наиболее распространенный способ сплайсинга у людей и высших эукариот), взаимоисключающие экзоны, альтернативные донорные или акцепторные сайты, удержание интронов (наиболее распространенный способ сплайсинга у растений, грибов и простейших), альтернативный сайт старта транскрипции (промотор) и альтернативное полиаденилирование.[101] Одна из целей RNA-Seq - выявить альтернативные события сплайсинга и проверить, различаются ли они в разных условиях. Секвенирование с длительным считыванием захватывает полный транскрипт и, таким образом, сводит к минимуму многие проблемы при оценке распространенности изоформ, такие как отображение неоднозначного чтения. Для коротко читаемой RNA-Seq существует несколько методов обнаружения альтернативного сплайсинга, которые можно разделить на три основные группы:[102][103][104]
- На основе подсчета (также на основе событий, дифференциальное соединение): оценить удержание экзонов. Примеры: DEXSeq,[105] КОВРИКИ,[106] и SeqGSEA.[107]
- На основе изоформы (также модули с множественным чтением, дифференциальное выражение изоформы): сначала оцените численность изоформ, а затем относительную численность между условиями. Примеры: Запонки 2[108] и DiffSplice.[109]
- На основе иссечения интрона: рассчитать альтернативное соединение с использованием разделенных чтений. Примеры: MAJIQ[110] и Leafcutter.[104]
Инструменты дифференциальной экспрессии генов также могут использоваться для дифференциальной экспрессии изоформ, если изоформы предварительно количественно определены с помощью других инструментов, таких как RSEM.[111]
Коэкспрессионные сети
Сети коэкспрессии - это основанные на данных представления генов, ведущих себя одинаково в разных тканях и в экспериментальных условиях.[112] Их основная цель заключается в генерировании гипотез и подходах по принципу «чувство вины по ассоциации» для определения функций ранее неизвестных генов.[112] Данные RNA-Seq были использованы для вывода генов, участвующих в конкретных путях, на основе Корреляции Пирсона, как в растениях[113] и млекопитающие.[114] Основное преимущество данных RNA-Seq в этом виде анализа по сравнению с платформами микрочипов - это способность покрывать весь транскриптом, что позволяет получить более полные представления о сетях регуляции генов.Дифференциальная регуляция изоформ сплайсинга одного и того же гена может быть обнаружена и использована для прогнозирования их биологических функций.[115][116] Сетевой анализ взвешенной коэкспрессии генов был успешно использован для идентификации модулей коэкспрессии и внутримодульных хаб-генов на основе данных последовательности РНК. Модули коэкспрессии могут соответствовать типам клеток или путям. Внутримодульные концентраторы с высокой степенью связи можно интерпретировать как представителей соответствующего модуля. Собственный ген - это взвешенная сумма экспрессии всех генов в модуле. Собственные гены - полезные биомаркеры (признаки) для диагностики и прогноза.[117] Были предложены подходы с преобразованием, стабилизирующим отклонения, для оценки коэффициентов корреляции на основе данных последовательности РНК.[113]
Вариант открытия
RNA-Seq фиксирует вариации ДНК, в том числе однонуклеотидные варианты, небольшие вставки / удаления. и структурная вариация. Вариант вызова in RNA-Seq аналогичен вызову вариантов ДНК и часто использует те же инструменты (включая SAMtools mpileup[118] и GATK HaplotypeCaller[119]) с поправками на сращивание. Одно уникальное измерение вариантов РНК - аллель-специфическая экспрессия (ASE): варианты только одного гаплотипа могут быть предпочтительно экспрессированы из-за регуляторных эффектов, включая печать и выражение количественных признаков локусов, и некодирование редкие варианты.[120][121] Ограничения идентификации варианта РНК включают то, что он отражает только экспрессируемые области (у людей <5% генома) и имеет более низкое качество по сравнению с прямым секвенированием ДНК.
Редактирование РНК (посттранскрипционные изменения)
Наличие совпадающих геномных и транскриптомных последовательностей человека может помочь обнаружить посттранскрипционные изменения (Редактирование РНК ).[3] Событие посттранскрипционной модификации идентифицируется, если транскрипт гена имеет аллель / вариант, не обнаруженный в геномных данных.
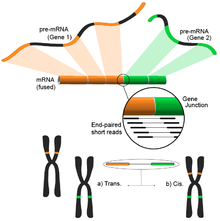
Обнаружение слитного гена
Вызванные различными структурными модификациями в геноме, гибридные гены привлекли внимание из-за их связи с раком.[122] Способность RNA-Seq беспристрастно анализировать весь транскриптом образца делает его привлекательным инструментом для поиска таких общих явлений при раке.[4]
Идея вытекает из процесса выравнивания коротких транскриптомных чтений с эталонным геномом. Большинство коротких прочтений попадают в один полный экзон, и ожидается, что меньший, но все же большой набор будет отображаться на известные экзон-экзонные соединения. Оставшиеся неотмеченные короткие считывания затем будут дополнительно проанализированы, чтобы определить, соответствуют ли они соединению экзон-экзон, где экзоны происходят от разных генов. Это свидетельствовало бы о возможном событии слияния, однако из-за продолжительности считывания это могло оказаться очень шумным. Альтернативный подход состоит в использовании чтения на конце пары, когда потенциально большое количество парных чтений будет отображать каждый конец на другой экзон, обеспечивая лучшее покрытие этих событий (см. Рисунок). Тем не менее, конечный результат состоит из множества и потенциально новых комбинаций генов, обеспечивающих идеальную отправную точку для дальнейшей проверки.
История

RNA-Seq была впервые разработана в середине 2000-е с появлением технологии секвенирования нового поколения.[123] Первые рукописи, которые использовали RNA-Seq даже без использования этого термина, включают рукописи рак простаты Сотовые линии[124] (от 2006 г.), Medicago truncatula[125] (2006), кукуруза[126] (2007), и Arabidopsis thaliana[127] (2007), а сам термин «RNA-Seq» впервые был упомянут в 2008 году.[128] Количество рукописей, ссылающихся на RNA-Seq в названии или аннотации (рисунок, синяя линия), постоянно увеличивается: в 2018 г. было опубликовано 6754 рукописи (ссылка на поиск PubMed ). Пересечение RNA-Seq и медицины (рисунок, золотая линия, ссылка на поиск PubMed ) имеет аналогичную скорость.[оригинальное исследование? ]
Приложения к медицине
RNA-Seq имеет потенциал для выявления новой биологии болезни, профильных биомаркеров для клинических показаний, определения путей воздействия лекарств и постановки генетического диагноза. Эти результаты могут быть дополнительно персонализированы для подгрупп или даже отдельных пациентов, потенциально подчеркивая более эффективную профилактику, диагностику и терапию. Осуществимость этого подхода частично диктуется денежными и временными затратами; Связанное с этим ограничение - это необходимая группа специалистов (биоинформатики, врачи / клиницисты, базовые исследователи, техники) для полной интерпретации огромного количества данных, полученных в результате этого анализа.[129]
Масштабные усилия по секвенированию
Большое внимание было уделено данным RNA-Seq после Энциклопедия элементов ДНК (ENCODE) и Атлас генома рака (TCGA) проекты использовали этот подход для характеристики десятков клеточных линий[130] и тысячи образцов первичных опухолей,[131] соответственно. ENCODE направлен на идентификацию регуляторных областей всего генома в различных когортах клеточных линий, и транскриптомные данные имеют первостепенное значение для понимания последующего эффекта этих эпигенетических и генетических регуляторных уровней. TCGA, напротив, нацелен на сбор и анализ тысяч образцов пациентов из 30 различных типов опухолей, чтобы понять основные механизмы злокачественной трансформации и прогрессирования. В этом контексте данные RNA-Seq предоставляют уникальный снимок транскриптомного статуса заболевания и позволяют рассматривать беспристрастную популяцию транскриптов, что позволяет идентифицировать новые транскрипты, гибридные транскрипты и некодирующие РНК, которые можно было бы не обнаружить с помощью различных технологий.
Смотрите также
Рекомендации
- ^ Шафи Т., Лоу Р. (2017). «Структура эукариотических и прокариотических генов». WikiJournal of Медицина. 4 (1). Дои:10.15347 / wjm / 2017.002.
- ^ Чу Й, Кори Д.Р. (август 2012 г.). «Секвенирование РНК: выбор платформы, экспериментальный дизайн и интерпретация данных». Нуклеиновые кислоты. 22 (4): 271–4. Дои:10.1089 / нат.2012.0367. ЧВК 3426205. PMID 22830413.
- ^ а б c Ван З., Герштейн М., Снайдер М. (январь 2009 г.). «RNA-Seq: революционный инструмент для транскриптомики». Обзоры природы. Генетика. 10 (1): 57–63. Дои:10.1038 / nrg2484. ЧВК 2949280. PMID 19015660.
- ^ а б Махер К.А., Кумар-Синха С., Цао Х, Кальяна-Сундарам С., Хан Б., Цзин Х и др. (Март 2009 г.). «Секвенирование транскриптома для обнаружения слияния генов при раке». Природа. 458 (7234): 97–101. Bibcode:2009Натура.458 ... 97М. Дои:10.1038 / природа07638. ЧВК 2725402. PMID 19136943.
- ^ Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (июль 2012 г.). «Стратегия профилирования рибосом для мониторинга трансляции in vivo путем глубокого секвенирования защищенных рибосомами фрагментов мРНК». Протоколы природы. 7 (8): 1534–50. Дои:10.1038 / nprot.2012.086. ЧВК 3535016. PMID 22836135.
- ^ Ли Дж. Х., Даугарти Э. Р., Шейман Дж., Калхор Р., Ян Дж. Л., Ферранте Т.С. и др. (Март 2014 г.). «Высоко мультиплексное секвенирование субклеточной РНК in situ». Наука. 343 (6177): 1360–3. Bibcode:2014Научный ... 343.1360L. Дои:10.1126 / science.1250212. ЧВК 4140943. PMID 24578530.
- ^ Кукурба К.Р., Монтгомери С.Б. (апрель 2015 г.). «Секвенирование и анализ РНК». Протоколы Колд-Спринг-Харбор. 2015 (11): 951–69. Дои:10.1101 / pdb.top084970. ЧВК 4863231. PMID 25870306.
- ^ а б c d е Гриффит М., Уокер-младший, Шпионы NC, Эйнскау Б.Дж., Гриффит О.Л. (август 2015 г.). «Информатика для секвенирования РНК: веб-ресурс для анализа в облаке». PLOS вычислительная биология. 11 (8): e1004393. Bibcode:2015PLSCB..11E4393G. Дои:10.1371 / journal.pcbi.1004393. ЧВК 4527835. PMID 26248053.
- ^ «РНК-секлопедия». rnaseq.uoregon.edu. Получено 2017-02-08.
- ^ Морин Р., Бейнбридж М., Фейес А., Херст М., Кшивински М., Пью Т. и др. (Июль 2008 г.). «Профилирование транскриптома HeLa S3 с использованием случайно примированной кДНК и массового параллельного секвенирования короткого чтения». Биотехнологии. 45 (1): 81–94. Дои:10.2144/000112900. PMID 18611170.
- ^ а б c Мортазави А., Уильямс Б.А., МакКью К., Шеффер Л., Уолд Б. (июль 2008 г.). «Картирование и количественная оценка транскриптомов млекопитающих с помощью RNA-Seq». Методы природы. 5 (7): 621–8. Дои:10.1038 / nmeth.1226. PMID 18516045. S2CID 205418589.
- ^ Chen EA, Souaiaia T, Herstein JS, Evgrafov OV, Spitsyna VN, Rebolini DF, Knowles JA (октябрь 2014 г.). «Влияние целостности РНК на однозначно картированные чтения в RNA-Seq». BMC Research Notes. 7 (1): 753. Дои:10.1186/1756-0500-7-753. ЧВК 4213542. PMID 25339126.
- ^ Лю Д., Грабер Дж. Х. (февраль 2006 г.). «Количественное сравнение библиотек EST требует компенсации систематических ошибок в генерации кДНК». BMC Bioinformatics. 7: 77. Дои:10.1186/1471-2105-7-77. ЧВК 1431573. PMID 16503995.
- ^ Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M и др. (Март 2018 г.). «Высоко параллельное прямое секвенирование РНК на массиве нанопор». Методы природы. 15 (3): 201–206. Дои:10.1038 / nmeth.4577. PMID 29334379. S2CID 3589823.
- ^ а б "Шапиро Э., Бизунер Т., Линнарссон С. (сентябрь 2013 г.). «Технологии, основанные на секвенировании отдельных клеток, произведут революцию в науке о целом организме». Обзоры природы. Генетика. 14 (9): 618–30. Дои:10.1038 / nrg3542. PMID 23897237. S2CID 500845."
- ^ Колодзейчик А.А., Ким Дж. К., Свенссон В., Мариони Дж. К., Тайхманн С.А. (май 2015 г.). «Технология и биология секвенирования одноклеточной РНК». Молекулярная клетка. 58 (4): 610–20. Дои:10.1016 / j.molcel.2015.04.005. PMID 26000846.
- ^ Монторо Д.Т., Хабер А.Л., Битон М., Винарский В., Лин Б., Биркет С.Е. и др. (Август 2018 г.). «Обновленная иерархия эпителия дыхательных путей включает ионоциты, экспрессирующие CFTR». Природа. 560 (7718): 319–324. Bibcode:2018Натура.560..319М. Дои:10.1038 / s41586-018-0393-7. ЧВК 6295155. PMID 30069044.
- ^ Плассхаерт Л.В., Жилионис Р., Чу-Винг Р., Савова В., Кнехр Дж., Рома Г. и др. (Август 2018 г.). «Одноклеточный атлас эпителия дыхательных путей показывает богатые CFTR легочные ионоциты». Природа. 560 (7718): 377–381. Bibcode:2018Натура.560..377П. Дои:10.1038 / s41586-018-0394-6. ЧВК 6108322. PMID 30069046.
- ^ Кляйн А.М., Мазутис Л., Акартуна I, Таллапрагада Н., Верес А., Ли В. и др. (Май 2015 г.). «Штрих-кодирование капель для транскриптомики одиночных клеток, применяемое к эмбриональным стволовым клеткам». Клетка. 161 (5): 1187–1201. Дои:10.1016 / j.cell.2015.04.044. ЧВК 4441768. PMID 26000487.
- ^ Макоско Э.З., Басу А., Сатия Р., Немеш Дж., Шекхар К., Голдман М. и др. (Май 2015 г.). «Профилирование экспрессии индивидуальных клеток с высокой степенью параллельности генома с использованием капель нанолитера». Клетка. 161 (5): 1202–1214. Дои:10.1016 / j.cell.2015.05.002. ЧВК 4481139. PMID 26000488.
- ^ "Hebenstreit D (ноябрь 2012 г.). "Методы, проблемы и возможности одноклеточной РНК-seq". Биология. 1 (3): 658–67. Дои:10.3390 / biology1030658. ЧВК 4009822. PMID 24832513."
- ^ Эбервин Дж., Сул Дж. Й., Бартфай Т., Ким Дж. (Январь 2014 г.). «Обещание секвенирования одной клетки». Методы природы. 11 (1): 25–7. Дои:10.1038 / nmeth.2769. PMID 24524134. S2CID 11575439.
- ^ Тан Ф., Барбачору С., Ван И, Нордман Э, Ли С., Сюй Н. и др. (Май 2009 г.). «Анализ целого транскриптома мРНК-Seq отдельной клетки». Методы природы. 6 (5): 377–82. Дои:10.1038 / NMETH.1315. PMID 19349980. S2CID 16570747.
- ^ Ислам С., Кьеллквист Ю., Молинер А., Заяк П., Фан Дж. Б., Лённерберг П., Линнарссон С. (июль 2011 г.). «Характеристика одноклеточного транскрипционного ландшафта с помощью высоко мультиплексной последовательности РНК». Геномные исследования. 21 (7): 1160–7. Дои:10.1101 / гр.110882.110. ЧВК 3129258. PMID 21543516.
- ^ Рамскельд Д., Луо С., Ван Ю.С., Ли Р., Дэн К., Фаридани О.Р. и др. (Август 2012 г.). «Полноразмерная мРНК-Seq из одноклеточных уровней РНК и отдельных циркулирующих опухолевых клеток». Природа Биотехнологии. 30 (8): 777–82. Дои:10.1038 / nbt.2282. ЧВК 3467340. PMID 22820318.
- ^ Хашимшони Т., Вагнер Ф, Шер Н., Янаи И. (сентябрь 2012 г.). «CEL-Seq: одноклеточная РНК-Seq путем мультиплексной линейной амплификации». Отчеты по ячейкам. 2 (3): 666–73. Дои:10.1016 / j.celrep.2012.08.003. PMID 22939981.
- ^ Сингх М., Аль-Эриани Дж., Карсуэлл С., Фергюсон Дж. М., Блэкберн Дж., Бартон К., Роден Д., Лучани Ф., Фан Т., Джунанкар С., Джексон К., Гуднау С.К., Смит М.А., Swarbrick A (2018). «Высокопроизводительное целевое долгосрочное секвенирование отдельных клеток выявляет клональный и транскрипционный ландшафт лимфоцитов». bioRxiv. Дои:10.1101/424945. PMID 31311926.
- ^ Сасагава Ю., Никайдо И., Хаяси Т., Данно Х., Уно К.Д., Имаи Т., Уэда Х.Р. (апрель 2013 г.). «Quartz-Seq: высоко воспроизводимый и чувствительный метод секвенирования одноклеточной РНК, выявляющий негенетическую гетерогенность экспрессии генов». Геномная биология. 14 (4): R31. Дои:10.1186 / gb-2013-14-4-r31. ЧВК 4054835. PMID 23594475.
- ^ Куно Т., Муди Дж., Квон А.Т., Шибаяма Ю., Като С., Хуанг И. и др. (Январь 2019). «C1 CAGE определяет сайты начала транскрипции и активность энхансера при разрешении одной клетки». Nature Communications. 10 (1): 360. Bibcode:2019НатКо..10..360K. Дои:10.1038 / s41467-018-08126-5. ЧВК 6341120. PMID 30664627.
- ^ Даль Молин А, Ди Камилло Б (2019). «Как разработать эксперимент по секвенированию одноклеточной РНК: подводные камни, проблемы и перспективы». Брифинги по биоинформатике. 20 (4): 1384–1394. Дои:10.1093 / bib / bby007. PMID 29394315.
- ^ Петерсон В.М., Чжан К.Х., Кумар Н., Вонг Дж., Ли Л., Уилсон, округ Колумбия, и др. (Октябрь 2017 г.). «Мультиплексное количественное определение белков и транскриптов в отдельных клетках». Природа Биотехнологии. 35 (10): 936–939. Дои:10.1038 / nbt.3973. PMID 28854175. S2CID 205285357.
- ^ Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. (Сентябрь 2017 г.). «Одновременное измерение эпитопа и транскриптома в отдельных клетках». Методы природы. 14 (9): 865–868. Дои:10.1038 / nmeth.4380. ЧВК 5669064. PMID 28759029.
- ^ Радж Б., Вагнер Д.Е., Маккенна А., Пандей С., Кляйн А.М., Шендур Дж. И др. (Июнь 2018). «Одновременное одноклеточное профилирование клонов и типов клеток в головном мозге позвоночных». Природа Биотехнологии. 36 (5): 442–450. Дои:10.1038 / nbt.4103. ЧВК 5938111. PMID 29608178.
- ^ Olmos D, Arkenau HT, Ang JE, Ledaki I, Attard G, Carden CP и др. (Январь 2009 г.). «Циркулирующие опухолевые клетки (ЦОК) считаются промежуточными конечными точками при устойчивом к кастрации раке простаты (CRPC): опыт одного центра». Анналы онкологии. 20 (1): 27–33. Дои:10.1093 / annonc / mdn544. PMID 18695026.
- ^ Левитин HM, Юань Дж., Sims PA (апрель 2018 г.). "Одноклеточный транскриптомный анализ неоднородности опухоли". Тенденции рака. 4 (4): 264–268. Дои:10.1016 / j.trecan.2018.02.003. ЧВК 5993208. PMID 29606308.
- ^ Джерби-Арнон Л., Шах П., Куоко М.С., Родман С., Су М.Дж., Мелмс Дж.С. и др. (Ноябрь 2018 г.). «Программа раковых клеток способствует исключению Т-клеток и устойчивости к блокаде контрольных точек». Клетка. 175 (4): 984–997.e24. Дои:10.1016 / j.cell.2018.09.006. ЧВК 6410377. PMID 30388455.
- ^ Стивенсон В., Донлин Л.Т., Батлер А., Розо С., Бракен Б., Рашидфаррохи А. и др. (Февраль 2018). «Последовательность одноклеточной РНК синовиальной ткани при ревматоидном артрите с использованием недорогих микрофлюидных инструментов». Nature Communications. 9 (1): 791. Bibcode:2018НатКо ... 9..791с. Дои:10.1038 / s41467-017-02659-х. ЧВК 5824814. PMID 29476078.
- ^ Avraham R, Haseley N, Brown D, Penaranda C, Jijon HB, Trombetta JJ и др. (Сентябрь 2015 г.). «Изменчивость патогена от клетки к клетке приводит к неоднородности иммунных ответов хозяина». Клетка. 162 (6): 1309–21. Дои:10.1016 / j.cell.2015.08.027. ЧВК 4578813. PMID 26343579.
- ^ Цао Дж., Пакер Дж. С., Рамани В., Кусанович Д. А., Хюин С., Даза Р. и др. (Август 2017 г.). «Комплексное одноклеточное транскрипционное профилирование многоклеточного организма». Наука. 357 (6352): 661–667. Bibcode:2017Научный ... 357..661C. Дои:10.1126 / science.aam8940. ЧВК 5894354. PMID 28818938.
- ^ Plass M, Solana J, Wolf FA, Ayoub S, Misios A, Glažar P и др. (Май 2018). «Атлас клеточного типа и древо родословной всего сложного животного с помощью одноклеточной транскриптомики». Наука. 360 (6391): eaaq1723. Дои:10.1126 / science.aaq1723. PMID 29674432.
- ^ Fincher CT, Wurtzel O, de Hoog T, Kravarik KM, Reddien PW (май 2018 г.). "Schmidtea mediterranea". Наука. 360 (6391): eaaq1736. Дои:10.1126 / science.aaq1736. ЧВК 6563842. PMID 29674431.
- ^ Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM (июнь 2018 г.). «Одноклеточное картирование ландшафтов экспрессии генов и клонов в эмбрионе рыбок данио». Наука. 360 (6392): 981–987. Bibcode:2018Sci ... 360..981W. Дои:10.1126 / science.aar4362. ЧВК 6083445. PMID 29700229.
- ^ Фаррелл Дж. А., Ван Й., Ризенфельд С. Дж., Шекхар К., Регев А., Шир А. Ф. (июнь 2018 г.). «Одноклеточная реконструкция траекторий развития во время эмбриогенеза рыбок данио». Наука. 360 (6392): eaar3131. Дои:10.1126 / science.aar3131. ЧВК 6247916. PMID 29700225.
- ^ Бриггс Дж. А., Вайнреб С., Вагнер Д. Е., Мегасон С., Пешкин Л., Киршнер М. В., Кляйн А. М. (июнь 2018 г.). «Динамика экспрессии генов в эмбриогенезе позвоночных при одноклеточном разрешении». Наука. 360 (6392): eaar5780. Дои:10.1126 / science.aar5780. ЧВК 6038144. PMID 29700227.
- ^ Ю Дж. «Научный прорыв 2018 года: отслеживание развития по ячейке». Научный журнал. Американская ассоциация развития науки.
- ^ а б Ли Х, Ловчи М.Т., Квон Ю.С., Розенфельд М.Г., Фу XD, Йео Г.В. (декабрь 2008 г.). «Определение плотности метки, необходимой для анализа цифрового транскриптома: приложение к андроген-чувствительной модели рака простаты». Труды Национальной академии наук Соединенных Штатов Америки. 105 (51): 20179–84. Bibcode:2008PNAS..10520179L. Дои:10.1073 / pnas.0807121105. ЧВК 2603435. PMID 19088194.
- ^ а б Stegle O, Parts L, Piipari M, Winn J, Durbin R (февраль 2012 г.). «Использование вероятностной оценки остатков экспрессии (PEER) для получения повышенной мощности и интерпретируемости анализов экспрессии генов». Протоколы природы. 7 (3): 500–7. Дои:10.1038 / nprot.2011.457. ЧВК 3398141. PMID 22343431.
- ^ Kingsford C, Patro R (июнь 2015 г.). «Сжатие коротких последовательностей на основе ссылок с использованием кодирования пути». Биоинформатика. 31 (12): 1920–8. Дои:10.1093 / биоинформатика / btv071. ЧВК 4481695. PMID 25649622.
- ^ а б Грабхерр М.Г., Хаас Б.Дж., Яссур М., Левин Дж.З., Томпсон Д.А., Амит И. и др. (Май 2011 г.). «Сборка полноразмерного транскриптома из данных RNA-Seq без эталонного генома». Природа Биотехнологии. 29 (7): 644–52. Дои:10.1038 / nbt.1883. ЧВК 3571712. PMID 21572440.
- ^ «Сборка De Novo с использованием чтения Illumina» (PDF). Получено 22 октября 2016.
- ^ Зербино Д.Р., Бирни Э. (май 2008 г.). "Velvet: алгоритмы сборки короткого чтения de novo с использованием графов де Брейна". Геномные исследования. 18 (5): 821–9. Дои:10.1101 / гр.074492.107. ЧВК 2336801. PMID 18349386.
- ^ Oases: ассемблер транскриптомов для очень коротких чтений
- ^ Чанг З., Ли Дж., Лю Дж., Чжан И, Эшби С., Лю Д. и др. (Февраль 2015 г.). «Бриджер: новая структура для сборки транскриптомов de novo с использованием данных RNA-seq». Геномная биология. 16 (1): 30. Дои:10.1186 / s13059-015-0596-2. ЧВК 4342890. PMID 25723335.
- ^ а б Ли Б., Филлмор Н., Бай И., Коллинз М., Томсон Дж. А., Стюарт Р., Дьюи С. Н. (декабрь 2014 г.). «Оценка сборок транскриптомов de novo по данным RNA-Seq». Геномная биология. 15 (12): 553. Дои:10.1186 / s13059-014-0553-5. ЧВК 4298084. PMID 25608678.
- ^ а б Добин А., Дэвис К.А., Шлезингер Ф., Дренкоу Дж., Залески С., Джа С. и др. (Январь 2013). «STAR: сверхбыстрый универсальный выравниватель RNA-seq». Биоинформатика. 29 (1): 15–21. Дои:10.1093 / биоинформатика / bts635. ЧВК 3530905. PMID 23104886.
- ^ Лэнгмид Б., Трапнелл С., Поп-М., Зальцберг С.Л. (2009). «Сверхбыстрое и эффективное с точки зрения памяти выравнивание коротких последовательностей ДНК с геномом человека». Геномная биология. 10 (3): R25. Дои:10.1186 / gb-2009-10-3-r25. ЧВК 2690996. PMID 19261174.
- ^ Трапнелл С., Пахтер Л., Зальцберг С.Л. (май 2009 г.). «TopHat: обнаружение сплайсинговых соединений с помощью RNA-Seq». Биоинформатика. 25 (9): 1105–11. Дои:10.1093 / биоинформатика / btp120. ЧВК 2672628. PMID 19289445.
- ^ Трапнелл С., Робертс А., Гофф Л., Пертеа Г., Ким Д., Келли Д. Р. и др. (Март 2012 г.). «Дифференциальный анализ экспрессии генов и транскриптов в экспериментах с последовательностью РНК с TopHat и Cufflinks». Протоколы природы. 7 (3): 562–78. Дои:10.1038 / nprot.2012.016. ЧВК 3334321. PMID 22383036.
- ^ Ляо И., Смит Г.К., Ши В. (май 2013 г.). «Выравниватель Subread: быстрое, точное и масштабируемое отображение чтения по принципу seed-and-voice». Исследования нуклеиновых кислот. 41 (10): e108. Дои:10.1093 / nar / gkt214. ЧВК 3664803. PMID 23558742.
- ^ Ким Д., Лангмид Б., Зальцберг С.Л. (апрель 2015 г.). «HISAT: выравниватель с быстрым сращиванием и малыми требованиями к памяти». Методы природы. 12 (4): 357–60. Дои:10.1038 / nmeth.3317. ЧВК 4655817. PMID 25751142.
- ^ Patro R, Mount SM, Kingsford C (май 2014 г.). «Sailfish обеспечивает количественную оценку изоформ без выравнивания по считыванию последовательности РНК с использованием легких алгоритмов». Природа Биотехнологии. 32 (5): 462–4. arXiv:1308.3700. Дои:10.1038 / nbt.2862. ЧВК 4077321. PMID 24752080.
- ^ Брей Н.Л., Пиментел Х, Мельстед П., Пахтер Л. (май 2016 г.). «Почти оптимальная вероятностная количественная оценка последовательности РНК». Природа Биотехнологии. 34 (5): 525–7. Дои:10.1038 / nbt.3519. PMID 27043002. S2CID 205282743.
- ^ Ву Т.Д., Ватанабэ СК (май 2005 г.). «GMAP: программа геномного картирования и выравнивания последовательностей мРНК и EST». Биоинформатика. 21 (9): 1859–75. Дои:10.1093 / биоинформатика / bti310. PMID 15728110.
- ^ Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR (февраль 2017 г.). «Комплексный сравнительный анализ выравнивателей RNA-seq на основе моделирования». Методы природы. 14 (2): 135–139. Дои:10.1038 / nmeth.4106. ЧВК 5792058. PMID 27941783.
- ^ Энгстрём П.Г., Штайгер Т., Сипос Б., Грант Г.Р., Калес А., Рэтш Г. и др. (Декабрь 2013). «Систематическая оценка программ сплайсированного выравнивания для данных RNA-seq». Методы природы. 10 (12): 1185–91. Дои:10.1038 / nmeth.2722. ЧВК 4018468. PMID 24185836.
- ^ Лу Б., Цзэн З., Ши Т. (февраль 2013 г.). «Сравнительное исследование сборки de novo и стратегий сборки под геномом для реконструкции транскриптома на основе RNA-Seq». Наука Китай Науки о жизни. 56 (2): 143–55. Дои:10.1007 / s11427-013-4442-z. PMID 23393030.
- ^ Брэднам К.Р., Фасс Дж. Н., Александров А., Баранай П., Бехнер М., Бирол И. и др. (Июль 2013). «Assemblathon 2: оценка de novo методов сборки генома у трех видов позвоночных». GigaScience. 2 (1): 10. arXiv:1301.5406. Bibcode:2013arXiv1301.5406B. Дои:10.1186 / 2047-217X-2-10. ЧВК 3844414. PMID 23870653.
- ^ Гринбаум Д., Коланджело С., Уильямс К., Герштейн М. (2003). «Сравнение содержания белка и уровней экспрессии мРНК в геномной шкале». Геномная биология. 4 (9): 117. Дои:10.1186 / gb-2003-4-9-117. ЧВК 193646. PMID 12952525.
- ^ Чжан Ж., Джавери Д. Д., Маршалл В. М., Бауэр Д. К., Эдсон Дж., Нараянан Р. К. и др. (Август 2014 г.). «Сравнительное исследование методов анализа дифференциальной экспрессии на данных RNA-Seq». PLOS ONE. 9 (8): e103207. Bibcode:2014PLoSO ... 9j3207Z. Дои:10.1371 / journal.pone.0103207. ЧВК 4132098. PMID 25119138.
- ^ Андерс С., Пил П. Т., Хубер В. (январь 2015 г.). «HTSeq - фреймворк Python для работы с высокопроизводительными данными секвенирования». Биоинформатика. 31 (2): 166–9. Дои:10.1093 / биоинформатика / btu638. ЧВК 4287950. PMID 25260700.
- ^ Ляо Ю., Смит Г.К., Ши В. (апрель 2014 г.). «featureCounts: эффективная программа общего назначения для сопоставления считываний последовательностей с геномными функциями». Биоинформатика. 30 (7): 923–30. arXiv:1305.3347. Дои:10.1093 / биоинформатика / btt656. PMID 24227677. S2CID 15960459.
- ^ Шмид М.В., Гроссниклаус У. (февраль 2015 г.). «Rcount: простой и гибкий подсчет чтения RNA-Seq». Биоинформатика. 31 (3): 436–7. Дои:10.1093 / биоинформатика / btu680. PMID 25322836.
- ^ Finotello F, Lavezzo E, Bianco L, Barzon L, Mazzon P, Fontana P и др. (2014). «Снижение систематической ошибки в данных секвенирования РНК: новый подход к подсчету». BMC Bioinformatics. 15 Дополнение 1 (Дополнение 1): S7. Дои:10.1186 / 1471-2105-15-s1-s7. ЧВК 4016203. PMID 24564404.
- ^ Хашимото ТБ, Эдвардс, доктор медицины, Гиффорд, округ Колумбия (март 2014 г.). «Универсальная коррекция счета для высокопроизводительного секвенирования». PLOS вычислительная биология. 10 (3): e1003494. Bibcode:2014PLSCB..10E3494H. Дои:10.1371 / journal.pcbi.1003494. ЧВК 3945112. PMID 24603409.
- ^ а б Робинсон, доктор медицины, Ошлак А (2010). «Метод масштабной нормализации для анализа дифференциальной экспрессии данных RNA-seq». Геномная биология. 11 (3): R25. Дои:10.1186 / gb-2010-11-3-r25. ЧВК 2864565. PMID 20196867.
- ^ Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ и др. (Май 2010 г.). «Сборка и количественная оценка транскриптов с помощью RNA-Seq выявляет неаннотированные транскрипты и переключение изоформ во время дифференцировки клеток». Природа Биотехнологии. 28 (5): 511–5. Дои:10.1038 / nbt.1621. ЧВК 3146043. PMID 20436464.
- ^ Пахтер Л. (19 апреля 2011 г.). «Модели для количественной оценки транскриптов из RNA-Seq». arXiv:1104.3889 [q-bio.GN ].
- ^ «Что такое FPKM? Обзор единиц экспрессии RNA-Seq». Фарраго. 8 мая 2014. Получено 28 марта 2018.
- ^ Вагнер Г.П., Кин К., Линч В.Дж. (декабрь 2012 г.). «Измерение количества мРНК с использованием данных RNA-seq: измерение RPKM несовместимо среди образцов». Теория в биологических науках = Theorie in den Biowissenschaften. 131 (4): 281–5. Дои:10.1007 / s12064-012-0162-3. PMID 22872506. S2CID 16752581.
- ^ Эванс, Кьяран; Хардин, Джоанна; Штобель, Даниэль М. (28 сентября 2018 г.). «Выбор методов нормализации RNA-Seq между образцами с точки зрения их предположений». Брифинги по биоинформатике. 19 (5): 776–792. Дои:10.1093 / bib / bbx008. ЧВК 6171491. PMID 28334202.
- ^ а б Ло CW, Чен Й, Ши В., Смит Г.К. (февраль 2014 г.). "voom: прецизионные веса разблокируют инструменты анализа линейной модели для счетчиков чтения RNA-seq". Геномная биология. 15 (2): R29. Дои:10.1186 / gb-2014-15-2-r29. ЧВК 4053721. PMID 24485249.
- ^ а б Андерс С., Хубер В. (2010). «Анализ дифференциальной экспрессии для данных подсчета последовательностей». Геномная биология. 11 (10): R106. Дои:10.1186 / gb-2010-11-10-r106. ЧВК 3218662. PMID 20979621.
- ^ а б Робинсон, доктор медицины, Маккарти, ди-джей, Смит, Г.К. (январь 2010 г.). «edgeR: пакет Bioconductor для анализа дифференциальной экспрессии цифровых данных экспрессии генов». Биоинформатика. 26 (1): 139–40. Дои:10.1093 / биоинформатика / btp616. ЧВК 2796818. PMID 19910308.
- ^ Маргерат С., Шмидт А., Кодлин С., Чен В., Эберсольд Р., Бэлер Дж. (Октябрь 2012 г.). «Количественный анализ транскриптомов и протеомов делящихся дрожжей в пролиферирующих и покоящихся клетках». Клетка. 151 (3): 671–83. Дои:10.1016 / j.cell.2012.09.019. ЧВК 3482660. PMID 23101633.
- ^ Оуэнс Н.Д., Блиц И.Л., Лейн М.А., Патрушев И., Овертон Д.Д., Гилкрист М.Дж. и др. (Январь 2016 г.). «Измерение абсолютного числа копий РНК при высоком временном разрешении выявляет кинетику транскриптома в развитии». Отчеты по ячейкам. 14 (3): 632–647. Дои:10.1016 / j.celrep.2015.12.050. ЧВК 4731879. PMID 26774488.
- ^ Ричи М.Э., Фипсон Б., Ву Д., Ху Ю., Ло С.В., Ши В., Смит Г.К. (апрель 2015 г.). "Limma поддерживает анализ дифференциальной экспрессии для секвенирования РНК и исследований микрочипов". Исследования нуклеиновых кислот. 43 (7): e47. Дои:10.1093 / nar / gkv007. ЧВК 4402510. PMID 25605792.
- ^ «Биокондуктор - программное обеспечение с открытым исходным кодом для биоинформатики».
- ^ Хубер В., Кэри В.Дж., Джентльмен Р., Андерс С., Карлсон М., Карвалью Б.С. и др. (Февраль 2015 г.). «Организация высокопроизводительного геномного анализа с помощью Bioconductor». Методы природы. 12 (2): 115–21. Дои:10.1038 / nmeth.3252. ЧВК 4509590. PMID 25633503.
- ^ Лик Дж. Т., Стори Дж. Д. (сентябрь 2007 г.). «Выявление гетерогенности в исследованиях экспрессии генов с помощью анализа суррогатных переменных». PLOS Genetics. 3 (9): 1724–35. Дои:10.1371 / journal.pgen.0030161. ЧВК 1994707. PMID 17907809.
- ^ Пиментел Х, Брей Н.Л., Пуэнте С., Мельстед П., Пахтер Л. (июль 2017 г.). «Дифференциальный анализ RNA-seq с учетом неопределенности количественного определения». Методы природы. 14 (7): 687–690. Дои:10.1038 / nmeth.4324. PMID 28581496. S2CID 15063247.
- ^ Трапнелл С., Хендриксон Д.Г., Соважо М., Гофф Л., Ринн Д.Л., Пахтер Л. (январь 2013 г.). «Дифференциальный анализ регуляции генов при разрешении транскриптов с помощью RNA-seq» (PDF). Природа Биотехнологии. 31 (1): 46–53. Дои:10.1038 / nbt.2450. ЧВК 3869392. PMID 23222703.
- ^ Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT (март 2015 г.). «Ballgown устраняет разрыв между сборкой транскриптома и анализом экспрессии». Природа Биотехнологии. 33 (3): 243–6. Дои:10.1038 / nbt.3172. ЧВК 4792117. PMID 25748911.
- ^ а б Сахрейян С.М., Мохиюддин М., Себра Р., Тилгнер Х., Афшар П.Т., Ау К.Ф. и др. (Июль 2017 г.). «Получение всестороннего биологического понимания транскриптома с помощью анализа РНК-секвенирования широкого спектра». Nature Communications. 8 (1): 59. Bibcode:2017НатКо ... 8 ... 59S. Дои:10.1038 / с41467-017-00050-4. ЧВК 5498581. PMID 28680106.
- ^ Ziemann M, Eren Y, El-Osta A (август 2016 г.). «Ошибки в названиях генов широко распространены в научной литературе». Геномная биология. 17 (1): 177. Дои:10.1186 / s13059-016-1044-7. ЧВК 4994289. PMID 27552985.
- ^ Soneson C, Delorenzi M (март 2013 г.). «Сравнение методов анализа дифференциальной экспрессии данных RNA-seq». BMC Bioinformatics. 14: 91. Дои:10.1186/1471-2105-14-91. ЧВК 3608160. PMID 23497356.
- ^ Фонсека Н.А., Мариони Дж., Бразма А. (30 сентября 2014 г.). «Профилирование генов RNA-Seq - систематическое эмпирическое сравнение». PLOS ONE. 9 (9): e107026. Bibcode:2014PLoSO ... 9j7026F. Дои:10.1371 / journal.pone.0107026. ЧВК 4182317. PMID 25268973.
- ^ Сейеднасроллах Ф., Лайхо А., Эло Л.Л. (январь 2015 г.). «Сравнение программных пакетов для определения дифференциальной экспрессии в исследованиях RNA-seq». Брифинги по биоинформатике. 16 (1): 59–70. Дои:10.1093 / bib / bbt086. ЧВК 4293378. PMID 24300110.
- ^ Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P и др. (2013). «Комплексная оценка методов дифференциального анализа экспрессии генов по данным RNA-seq». Геномная биология. 14 (9): R95. Дои:10.1186 / gb-2013-14-9-r95. ЧВК 4054597. PMID 24020486.
- ^ Конеса А., Мадригал П., Таразона С., Гомес-Кабреро Д., Сервера А., Макферсон А. и др. (Январь 2016 г.). «Обзор лучших практик анализа данных RNA-seq». Геномная биология. 17 (1): 13. Дои:10.1186 / s13059-016-0881-8. ЧВК 4728800. PMID 26813401.
- ^ Коста-Силва Дж., Домингес Д., Лопес FM (21 декабря 2017 г.). «Анализ дифференциальной экспрессии RNA-Seq: расширенный обзор и программный инструмент». PLOS ONE. 12 (12): e0190152. Bibcode:2017PLoSO..1290152C. Дои:10.1371 / journal.pone.0190152. ЧВК 5739479. PMID 29267363.
- ^ а б Керен Х., Лев-Маор Г., Аст Г. (май 2010 г.). «Альтернативный сплайсинг и эволюция: диверсификация, определение экзона и функция». Обзоры природы. Генетика. 11 (5): 345–55. Дои:10.1038 / nrg2776. PMID 20376054. S2CID 5184582.
- ^ Лю Р., Лорейн А.Э., Дикерсон Дж. А. (декабрь 2014 г.). «Сравнение вычислительных методов для дифференциального обнаружения альтернативного сплайсинга с использованием RNA-seq в растительных системах». BMC Bioinformatics. 15 (1): 364. Дои:10.1186 / s12859-014-0364-4. ЧВК 4271460. PMID 25511303.
- ^ Пахтер, Лиор (19 апреля 2011 г.). «Модели для количественной оценки транскриптов из RNA-Seq». arXiv:1104.3889 [q-bio.GN ].
- ^ а б Ли Ю.И., Ноулз Д.А., Хамфри Дж., Барбейра А.Н., Дикинсон С.П., Им Г.К., Причард Дж. К. (январь 2018 г.). «Количественная оценка сплайсинга РНК без аннотаций с использованием LeafCutter». Природа Генетика. 50 (1): 151–158. Дои:10.1038 / s41588-017-0004-9. ЧВК 5742080. PMID 29229983.
- ^ Андерс С., Рейес А., Хубер В. (октябрь 2012 г.). «Обнаружение дифференциального использования экзонов из данных RNA-seq». Геномные исследования. 22 (10): 2008–17. Дои:10.1101 / гр.133744.111. ЧВК 3460195. PMID 22722343.
- ^ Шен С., Пак Дж. У., Хуанг Дж., Дитмар К. А., Лу З. X, Чжоу К. и др. (Апрель 2012 г.). «MATS: байесовская структура для гибкого обнаружения дифференциального альтернативного сплайсинга на основе данных RNA-Seq». Исследования нуклеиновых кислот. 40 (8): e61. Дои:10.1093 / nar / gkr1291. ЧВК 3333886. PMID 22266656.
- ^ Ван X, Кэрнс MJ (июнь 2014 г.). «SeqGSEA: пакет Bioconductor для анализа обогащения набора генов данных РНК-Seq, интегрирующий дифференциальную экспрессию и сплайсинг». Биоинформатика. 30 (12): 1777–9. Дои:10.1093 / биоинформатика / btu090. PMID 24535097.
- ^ Трапнелл С., Хендриксон Д.Г., Соважо М., Гофф Л., Ринн Д.Л., Пахтер Л. (январь 2013 г.). «Дифференциальный анализ регуляции генов при разрешении транскриптов с помощью RNA-seq». Природа Биотехнологии. 31 (1): 46–53. Дои:10.1038 / nbt.2450. ЧВК 3869392. PMID 23222703.
- ^ Ху Y, Хуанг Y, Du Y, Orellana CF, Singh D, Johnson AR, et al. (Январь 2013). «DiffSplice: обнаружение в масштабе всего генома событий дифференциального сплайсинга с помощью RNA-seq». Исследования нуклеиновых кислот. 41 (2): e39. Дои:10.1093 / нар / гкс1026. ЧВК 3553996. PMID 23155066.
- ^ Вакеро-Гарсия Дж., Баррера А., Газзара М. Р., Гонсалес-Валлинас Дж., Лахенс Н. Ф., Хогенеш Дж. Б. и др. (Февраль 2016). «Новый взгляд на сложность и регуляцию транскриптома через призму локальных вариаций сплайсинга». eLife. 5: e11752. Дои:10.7554 / eLife.11752. ЧВК 4801060. PMID 26829591.
- ^ Мерино Г.А., Конеса А., Фернандес Е.А. (март 2019 г.). «Сравнительный анализ рабочих процессов для обнаружения дифференциального сплайсинга и дифференциальной экспрессии на уровне изоформ в исследованиях человеческих последовательностей РНК». Брифинги по биоинформатике. 20 (2): 471–481. Дои:10.1093 / нагрудник / bbx122. PMID 29040385. S2CID 22706028.
- ^ а б Маркотт Э.М., Пеллегрини М., Томпсон М.Дж., Йейтс Т.О., Айзенберг Д. (ноябрь 1999 г.). «Комбинированный алгоритм для геномного предсказания функции белка». Природа. 402 (6757): 83–6. Bibcode:1999Натура 402 ... 83М. Дои:10.1038/47048. PMID 10573421. S2CID 144447.
- ^ а б Giorgi FM, Del Fabbro C, Licausi F (март 2013 г.). «Сравнительное исследование сетей коэкспрессии на основе РНК-секвенций и микрочипов у Arabidopsis thaliana». Биоинформатика. 29 (6): 717–24. Дои:10.1093 / биоинформатика / btt053. PMID 23376351.
- ^ Янку О.Д., Каван С., Нижний Д., Сирлз Р., Хитземанн Р., МакВини С. (июнь 2012 г.). «Использование данных RNA-Seq для вывода сети de novo coexpression». Биоинформатика. 28 (12): 1592–7. Дои:10.1093 / биоинформатика / bts245. ЧВК 3493127. PMID 22556371.
- ^ Экси Р., Ли ХД, Менон Р., Вэнь Й., Оменн Г.С., Кретцлер М., Гуань Й. (ноябрь 2013 г.). «Систематическая дифференциация функций для альтернативно сплайсированных изоформ посредством интеграции данных RNA-seq». PLOS вычислительная биология. 9 (11): e1003314. Bibcode:2013PLSCB ... 9E3314E. Дои:10.1371 / journal.pcbi.1003314. ЧВК 3820534. PMID 24244129.
- ^ Ли HD, Менон Р., Оменн Г.С., Гуань Й. (август 2014 г.). «Наступающая эра интеграции геномных данных для анализа функции изоформ сплайсинга». Тенденции в генетике. 30 (8): 340–7. Дои:10.1016 / j.tig.2014.05.005. ЧВК 4112133. PMID 24951248.
- ^ Форушани А., Аграхари Р., Докинг Р., Чанг Л., Дунс Г., Худоба М. и др. (Март 2017 г.). «Масштабный анализ генной сети показывает важность пути внеклеточного матрикса и генов гомеобокса при остром миелоидном лейкозе: введение в пакет Pigengene и его приложения». BMC Medical Genomics. 10 (1): 16. Дои:10.1186 / s12920-017-0253-6. ЧВК 5353782. PMID 28298217.
- ^ Ли Х, Хандакер Б., Вайсокер А., Феннелл Т., Руан Дж., Гомер Н. и др. (Август 2009 г.). "Формат выравнивания / карты последовательностей и SAMtools". Биоинформатика. 25 (16): 2078–9. Дои:10.1093 / биоинформатика / btp352. ЧВК 2723002. PMID 19505943.
- ^ ДеПристо М.А., Бэнкс Э., Поплин Р., Гаримелла К.В., Магуайр Дж. Р., Хартл С. и др. (Май 2011 г.). «Основа для обнаружения вариаций и генотипирования с использованием данных секвенирования ДНК следующего поколения». Природа Генетика. 43 (5): 491–8. Дои:10,1038 / нг.806. ЧВК 3083463. PMID 21478889.
- ^ Battle A, Brown CD, Engelhardt BE, Montgomery SB (октябрь 2017 г.). «Генетические эффекты на экспрессию генов в тканях человека». Природа. 550 (7675): 204–213. Bibcode:2017Натура.550..204А. Дои:10.1038 / природа24277. ЧВК 5776756. PMID 29022597.
- ^ Richter F, Hoffman GE, Manheimer KB, Patel N, Sharp AJ, McKean D, et al. (Март 2019 г.). «ORE определяет экстремальные эффекты экспрессии, обогащенные для редких вариантов». Биоинформатика. 35 (20): 3906–3912. Дои:10.1093 / биоинформатика / btz202. ЧВК 6792115. PMID 30903145.
- ^ Тейшейра MR (декабрь 2006 г.). «Рецидивирующие онкогены слияния при карциномах». Критические обзоры онкогенеза. 12 (3–4): 257–71. Дои:10.1615 / critrevoncog.v12.i3-4.40. PMID 17425505.
- ^ Вебер А.П. (ноябрь 2015 г.). «Открытие новой биологии посредством секвенирования РНК». Физиология растений. 169 (3): 1524–31. Дои:10.1104 / стр.15.01081. ЧВК 4634082. PMID 26353759.
- ^ Бейнбридж М.Н., Уоррен Р.Л., Херст М., Романюк Т., Зенг Т., Го А. и др. (Сентябрь 2006 г.). «Анализ транскриптома клеточной линии рака простаты LNCaP с использованием подхода секвенирования путем синтеза». BMC Genomics. 7: 246. Дои:10.1186/1471-2164-7-246. ЧВК 1592491. PMID 17010196.
- ^ Cheung F, Haas BJ, Goldberg SM, May GD, Xiao Y, Town CD (октябрь 2006 г.). «Секвенирование экспрессированных секвенированных тегов Medicago truncatula с использованием технологии 454 Life Sciences». BMC Genomics. 7: 272. Дои:10.1186/1471-2164-7-272. ЧВК 1635983. PMID 17062153.
- ^ Emrich SJ, Barbazuk WB, Li L, Schnable PS (январь 2007 г.). «Открытие и аннотация генов с использованием секвенирования транскриптома LCM-454». Геномные исследования. 17 (1): 69–73. Дои:10.1101 / гр.5145806. ЧВК 1716268. PMID 17095711.
- ^ Вебер А.П., Вебер К.Л., Карр К., Вилкерсон С., Ольрогге Дж. Б. (май 2007 г.). «Отбор образцов транскриптома Arabidopsis с массовым параллельным пиросеквенированием». Физиология растений. 144 (1): 32–42. Дои:10.1104 / стр. 107.096677. ЧВК 1913805. PMID 17351049.
- ^ Нагалакшми Ю., Ван З., Верн К., Шоу С., Раха Д., Герштейн М., Снайдер М. (июнь 2008 г.). «Транскрипционный ландшафт генома дрожжей, определенный с помощью секвенирования РНК». Наука. 320 (5881): 1344–9. Bibcode:2008Научный ... 320.1344N. Дои:10.1126 / science.1158441. ЧВК 2951732. PMID 18451266.
- ^ Сандберг, Рикард (30 декабря 2013 г.). «Вступление в эру одноклеточной транскриптомики в биологии и медицине». Методы природы. 11 (1): 22–24. Дои:10.1038 / nmeth.2764. ISSN 1548-7091.
- ^ «КОДИРОВАТЬ матрицу данных». Получено 2013-07-28.
- ^ "Атлас генома рака - портал данных". Получено 2013-07-28.
внешняя ссылка
| Scholia имеет тема профиль для РНК-Seq. |
- RNA-Seq для всех: руководство высокого уровня по разработке и реализации эксперимента RNA-Seq.
- Тагучи, Ю.-х. (2019). «Сравнительный анализ транскриптомики». Энциклопедия биоинформатики и вычислительной биологии. С. 814–818. Дои:10.1016 / B978-0-12-809633-8.20163-5. ISBN 9780128114322.
- Справочный модуль по естественным наукам