Сокращение березы - Birch reduction
| Сокращение березы | |
|---|---|
| Названный в честь | Артур Берч |
| Тип реакции | Органическая окислительно-восстановительная реакция |
| Идентификаторы | |
| Портал органической химии | березовый |
| RSC ID онтологии | RXNO: 0000042 |
В Сокращение березы это органическая реакция, которая используется для преобразования арены к циклогексадиены. Реакция названа в честь австралийского химика. Артур Берч. В этом органическое восстановление из ароматические кольца в жидкости аммиак с участием натрий, литий, или калий и алкоголь, такие как этиловый спирт и терт-бутанол. Эта реакция не похожа на каталитический гидрирование, который обычно полностью восстанавливает ароматическое кольцо до циклогексан.[1][2]

Примером может служить сокращение нафталин:[3]

Основной механизм реакции
Раствор натрия в жидком аммиаке состоит из электрид соль [Na (NH3)Икс]+ е−, который имеет очень интенсивный синий цвет. В сольватированные электроны добавить к ароматическому кольцу, чтобы анион-радикал. Добавленный спирт поставляет протон к анион-радикалу, а также к предпоследнему карбаниону; для большинства субстратов аммиак недостаточно кислый.[4]

Региоселективность
Восстановление анизола является одним из простейших примеров и показано в уравнении 1. Восстановление бензойной кислоты показано в уравнении 2.
Место на ринге, где анион-радикал изначально протонирован, определяет структуру продукта. При использовании донора электронов, такого как метокси (MeO), протонирование алкила некоторыми исследователями рассматривается как орто (т.е. рядом или 1,2) к заместителю. Другие исследователи полагали, что протонирование мета (1,3) к заместителю. Артур Берч одобрил мета протонирование. Считается, что с электроноакцепторными заместителями протонирование происходит в месте заместителя (ipso) или параграф (1,4), но это тоже неясно. Эмпирические правила А. Дж. Берча гласят, что для донорных заместителей конечный продукт имеет максимальное количество заместителей на конечных двойных связях. Для электроноакцепторных групп двойные связи продукта избегают заместителей. Предпочтение размещения групп во время реакции и в конечном продукте называется региоселективностью.


Общие детали механизма реакции
Раствор металла в аммиаке обеспечивает электроны которые захватываются ароматическим кольцом с образованием соответствующего анион-радикала B на первой стадии реакции. За этим следует протонирование спиртом с образованием циклогексадиенильного радикала C. Затем второй электрон переносится на радикал с образованием циклогексадиенилкарбаниона D. На последней стадии второй протон приводит циклогексадиенильный карбанион к неконъюгированный циклогексадиенильный продукт. Эти шаги описаны ниже для анизола.

Известно, что реакция третий порядок - первый порядок по ароматическим, первый по щелочному металлу и первый по спирту.[5] Для этого необходимо, чтобы ограничивающий шаг быть превращением анион-радикала B в циклогексадиенильный радикал C.
Региоселективность реакции
Редукция Березы имеет несколько сложных механистических особенностей. Эти особенности определяют реакцию региоселективность и рассматриваются ниже. Правило Берча для ароматических соединений с донорами электронов, такими как метоксил или алкил, заключается в том, что продукт будет иметь остаточные двойные связи, несущие максимальное количество заместителей. Для ароматических углеводородов с электроноакцепторными группами, такими как карбоксил, группы заместителей избегают двойных связей. В обоих случаях, с донорскими и отводящими группами, остаточные двойные связи неконъюгированы (см. Ниже). Механизмы реакции, объясняющие эту региоселективность, представляют большой научный интерес. Основные особенности:
- В жидком аммиаке щелочные металлы растворяются с образованием синего раствора, который упрощенно рассматривается как имеющий "свободные электроны ". Электроны захватываются ароматическим кольцом, по одному. После того, как первый электрон поглощается, образуется анион-радикал. Затем молекула спирта отдает свой гидроксильный водород, чтобы сформировать новый Связь C – H; на данный момент радикальный был сформирован. За этим следует захват второго электрона, карбанион типа циклогексадиенила (т.е. с C = C – C – C = C в шестичленном кольце с отрицательным зарядом). Тогда этот циклогексадиенильный анион является протонированный присутствующим алкоголем. Протонирование происходит в центре циклогексадиенильной системы. Эта (регио-) избирательность характерна.
- То, где изначально протонирован анион-радикал, определяет структуру продукта. С донором электронов, таким как метокси (MeO), или с алкильной группой, некоторые исследователи считали протонирование орто (т.е. рядом или 1,2) к заместителю. Другие исследователи полагали, что протонирование мета (1,3) к заместителю. Артур Берч одобрил мета протонирование. Считалось, что с электроноакцепторными заместителями протонирование происходит в месте заместителя (ipso) или параграф (1,4). Опять же, мнения разделились. Эмпирические правила А. Дж. Берча гласят, что для донорных заместителей конечный продукт имеет максимальное количество заместителей на конечных двойных связях. Для электроноакцепторных групп двойные связи продукта избегают заместителей. Предпочтение размещения групп в механизме и в конечном продукте называется региоселективностью.
- Механизм реакции предоставляет подробную информацию о молекулярном изменении по мере протекания реакции. В случае благотворительных групп предпочтение А. Дж. Берча мета Протонирование анион-радикала было основано на качественных соображениях, но экспериментально это не было продемонстрировано.
- В 1961 году простой расчет электронной плотности анион-радикала показал, что это орто сайт, который был наиболее отрицательным и, следовательно, с наибольшей вероятностью протонировал. Кроме того, с помощью вычислений было определено, что второе протонирование происходит в центре циклогексадиенильного аниона с образованием неконъюгированного продукта.
- Неопределенность в химической литературе теперь имеет только историческое значение. Действительно, сообщалось о некоторых дополнительных результатах вычислений, которые не предполагают предпочтения мета радикально-анионного протонирования до смеси орто и мета протонирование.[нужна цитата ]
- В 1990 и 1993 годах был разработан эзотерический тест, который показал, что орто протонирование анион-радикала было предпочтительнее, чем мета (семь к одному).[нужна цитата ] Это сопровождалось более современными вычислениями, которые совпали. И эксперимент, и расчеты соответствовали расчетам начала 1961 года.
- В случае электроноакцепторных групп в литературе есть примеры, демонстрирующие природу карбаниона непосредственно перед окончательным протонированием,[нужна цитата ] обнаруживая, что протонирование первичного анион-радикала происходит параграф к отходящему заместителю.
- Остается обсудить окончательное протонирование циклогексадиенильного аниона. В 1961 году было обнаружено, что простые вычисления Хюккеля не позволяют различить различные места протонирования.[нужна цитата ] Однако, когда вычисления были изменены с несколько более реалистичными предположениями, вычисления Хюккеля показали, что центральный углерод является предпочтительным. Более современные вычисления 1990 и 1993 гг. Согласуются.[нужна цитата ]
Механизм
Механизм редукции березы был предметом многочисленных дискуссий. Первоначальный механизм восстановления по Берче предполагал протонирование анион-радикала, который был мета с метокси и алкильными группами кольца. Он также предположил, что последняя стадия, протонирование аниона циклогексадиенила, происходила орто по отношению к этим заместителям. Первоначальный механизм Берча был основан на качественных рассуждениях, а именно на том, что анион-радикальный электронная плотность в результате добавления электрона будет наивысшим мета к донору электронов (такому как метокси или метил) из-за избегания обычных орто-параграф высокая плотность у нейтральных видов.[6]
В 1961 году простой Hückel Расчеты показали, что предложенный Берчем механизм неверен. Правильный механизм O изображен ниже.[7][8]Два априорных альтернативных механизма O и M:
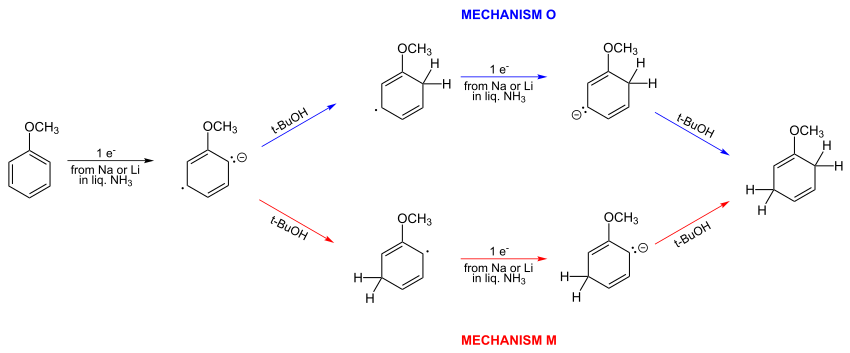
Берч не согласился с этим выводом и продолжил предлагать мета протонирование анион-радикала. Он предложил мета атака является результатом "противодействия орто и параграф начальный заряд ».[9] Ботнер-Би в 1959 г. привел качественные аргументы в пользу мета-протонирование[5] как было предложено ранее Берчем.
Бернхэм в 1969 году пришел к выводу, что протонирование маловероятно преимущественно на орто позиции и реакция, скорее всего, происходит в мета положение, но может происходить на обоих сайтах с одинаковой скоростью.[10]
Впоследствии Берч в обзорной статье[11] отметил, что в то время не существовало экспериментального метода, который позволил бы определить правильный механизм. Но он заметил, что публикация Бернхема[10] одобренный мета атака.
В публикациях 1980 г. Берч сотрудничал с Лев Радом в исследовании, которое пришло к выводу, что концентрации электронов на орто и мета позиции для закрытия с небольшим орто предпочтение, но со смесями орто и мета происходит протонирование.[12][13] Ограниченный Метод Хартри-Фока на орбитальный слейтер (3-g), и неограниченная орбитальная орбиталь типа Хартри-Фока на одном и том же базисном наборе, были использованы для заключения, что оба орто и мета замены будут происходить с небольшим предпочтением орто.[12][13]
Экспериментальное тестирование и вычислительная проверка
Затем в 1990 и 1993 годах был наконец разработан метод экспериментальной оценки того, протонировали ли анизол и анион-радикал толуола. орто или мета.[14][15] Эзотерический метод начался с предпосылки, что изотопная селективность при протонировании в протий-дейтериевой среде будет больше для анион-радикала первой стадии протонирования, чем для карбаниона предпоследней стадии. Причина заключалась в том, что карбанионы являются гораздо более основными, чем соответствующие анион-радикалы, и поэтому будут реагировать более экзотермически и менее избирательно при протонировании. Экспериментально было установлено, что меньше дейтерия в орто сайт чем мета результат (1: 7) для различных метоксилированных ароматических углеводородов. Это следствие большей селективности протонирования анион-радикала. Вычисления (например, ROHF / 6-31g) электронных плотностей совпали с экспериментальными наблюдениями. Также было установлено, что приграничные орбитальные плотности не совпадают, и они использовались в некоторых предыдущих отчетах.
Впоследствии, в 1992 и 1996 годах, Birch дважды публиковался, хотя мета протонирование было предпочтительным.[16][17] Это было изменением его более ранних взглядов, опубликованных вместе с Лео Радомом.
Однако в учебниках, посвященных механизму березового сокращения, отмечается, что орто протонирование исходного анион-радикала является предпочтительным.[18]
Восстановление березы электроноакцепторными заместителями
В отличие от примеров с электронодонорными заместителями, случай с акцептирующими группами более очевиден. Таким образом, как показано ниже, структура предпоследнего дианиона D характеризуется тем, что он подвержен улавливанию алкилгалогенидами.
Механизм восстановления бензойных кислот, включая возможное алкилирование

Этот дианион возникает независимо от того, используется ли алкоголь в восстановлении или нет. Таким образом, начальное протонирование терт-бутиловый спирт или аммиак параграф а не ipso, как видно на этапе от B до C.[19][20][21]
Второй этап восстановления березы с региохимией с образованием неконъюгированных циклогексадиенов.
Второй шаг редукции березы, дающий неконъюгированный cyclohexadienes также ставит механистические вопросы. Таким образом, как показано на рисунке ниже, для карбаниона существуют три резонансные структуры B, C и D. Простые вычисления Хюккеля приводят, как отмечено в первой записи таблицы ниже, к равным электронным плотностям у трех атомов 1, 3 и 5. Однако, в отличие от плотностей, расчет Хюккеля менее наивен относительно поручения на облигации,[7][22][23] а облигации 2–3 и 5–6 будут сокращены, как показано в первой записи таблицы. С помощью порядков связи, изменяющих простые обменные интегралы, в вычислении Малликена-Веланда-Манна было показано, что плотность электронов в центральном атоме 1 становится наибольшей.[22][23] Более современные вычисления RHF приводят к тому же результату.[14][15]
Введение электрона в бензол и 3 резонансные структуры для карбаниона второй стадии и центральное протонирование с образованием неконъюгированного диена:

Пять атомов углерода циклогексадиенильного аниона.[22][23]
| Приближение | Плотность атома 3 | Плотность Атом 2 | Плотность атома 1 | Приказ на облигации 2–3 | Приказ на облигации 1–2 |
|---|---|---|---|---|---|
| Хюккель (1-е прибл.) | 0.333 | 0.00 | 0.333 | 0.788 | 0.578 |
| 2-й ок. | 0.317 | 0.00 | 0.365 | 0.802 | 0.564 |
| 3-й ок. | 0.316 | 0.00 | 0.368 | 0.802 | 0.562 |
Известны прецеденты протонирования центрального аниона.[7][24] Таким образом, конъюгированные еноляты как C = CC = CO- в течение некоторого времени были известны как кинетически протонирующие в центре енолятной системы с образованием β, γ-ненасыщенного карбонильного соединения в условиях, когда анион, а не енол, является разновидностью протонированный.
Березовое алкилирование
При наличии алкилгалогенид то карбанион также может пройти нуклеофильное замещение с участием углерод-углеродная связь формирование. В замещенных ароматических соединениях электроноакцепторный заместитель, например карбоновая кислота,[25] стабилизирует карбанион и наименее замещенные олефин генерируется. С электронодонорный заместитель получается обратный эффект.[26] В результате реакции образуется больше менее термодинамически стабильного несопряженного 1,4-аддитивного продукта, чем более стабильного сопряженный 1,3-диен, потому что наибольший орбитальный коэффициент HOMO сопряженного промежуточного пентадиенильного аниона находится на центральном атоме углерода. После образования образующийся 1,4-циклогексадиен не может уравновеситься с термодинамически более стабильным продуктом; следовательно, получается наблюдаемый кинетический продукт. Более безопасные экспериментальные альтернативы щелочным металлам, такие как Восстановитель M-SG, тоже существуют.
В Березовое алкилирование то анион образующийся в редукции березы улавливается подходящим электрофил например, галогеналкан, Например:[27]

В реакции, изображенной ниже, 1,4-дибромбутан добавляют к терт-бутилбензоат с образованием алкилированного 1,4-циклогексадиенового продукта:[28]

Модификации
Поскольку жидкий аммиак должен конденсироваться в колбе и испаряться в течение ночи после завершения реакции, вся процедура может быть довольно хлопотной и трудоемкой. Однако использовались альтернативные растворители, такие как THF[29][30] а также смесь п-пропиламин и этилендиамин,[31] оба с сопоставимыми результатами. Последний на самом деле является модификацией Реакция Бенкесера, который в своей первоначальной форме имеет тенденцию полностью восстанавливать нафталин до октагидро- и декагидронафталина.
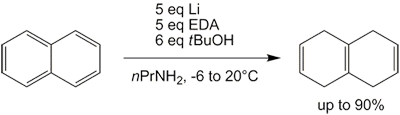
Это восстановление нафталина до изотетралина (1,4,5,8-тетрагидронафталина) дает некоторые тетралин (1,2,3,4-тетрагидронафталин) в качестве побочного продукта, как и в случае обычного восстановления по Берчу.
Восстановление может осуществляться за счет внешнего потенциала жертвенного анода (магния или алюминия). Диметилмочевина, донор протонов, стабилизирует интермедиат ионами лития (из солей). [32]
История
Реакция приписывается Артур Берч (1915–1995) во время работы в Лаборатория Дайсона Перринса на Оксфордский университет,[6][33][34][35][36][37] опираясь на более ранние работы Вустера и Годфри, опубликованные в 1937 году.[38] Он преобразует ароматические соединения иметь бензоид кольцо в продукт, 1,4-циклогексадиены, в котором два атома водорода присоединены к противоположным концам молекулы.
Первоначальная реакция, описанная Берчем в 1944 г., использовала натрий и этиловый спирт.[6][33][34] Альфред Л. Уайлдс позже обнаружил, что литий дает лучшие урожаи.[39][40]
Дополнительное чтение
- Кейн, Д. (1976). «Восстановление и родственные реакции α, β-ненасыщенных карбонильных соединений с металлами в жидком аммиаке». Орг. Реагировать. (обзор). 23: 1–258. Дои:10.1002 / 0471264180.or023.01. ISBN 0471264180.
Смотрите также
использованная литература
- ^ Rabideau, P.W .; Марцинов, З. (1992). «Березовое восстановление ароматических соединений». Орг. Реагировать. (обзор). 42: 1–334. Дои:10.1002 / 0471264180.or042.01. ISBN 0471264180.
- ^ Мандер, Л. Н. (1991). «Частичное восстановление ароматических колец растворением металлов и другими методами». Компр. Орг. Synth. (обзор). 8: 489–521. Дои:10.1016 / B978-0-08-052349-1.00237-7. ISBN 978-0-08-052349-1.
- ^ Vogel, E .; Klug, W .; Брейер, А. (1974). «1,6-Метано [10] аннулен». Органический синтез.; Коллективный объем, 6
- ^ Марш, Джерри (1985), Продвинутая органическая химия: реакции, механизмы и структура (3-е изд.), Нью-Йорк: Wiley, ISBN 0-471-85472-7
- ^ а б Крапчо, А. П .; Ботнер-Би, А.А. (1959). "Кинетика восстановления бензола и замещенных бензолов металл-аммиак-спирт1". Варенье. Chem. Soc. 81 (14): 3658–3666. Дои:10.1021 / ja01523a042.
- ^ а б c Берч, А. Дж. (1944). «Восстановление растворением металлов. Часть I». J. Chem. Soc.: 430. Дои:10.1039 / JR9440000430.
- ^ а б c Циммерман, Х. Э. (1961). «Ориентация в восстановлении металлического аммиака». Тетраэдр. 16 (1–4): 169–176. Дои:10.1016/0040-4020(61)80067-7.
- ^ «Катализируемые основаниями перегруппировки», глава 6 «Молекулярных перегруппировок», Циммерман, Х. Э., под ред. П. ДеМайо, Interscience, 345–406, Нью-Йорк, 1963.
- ^ Берч, А. Дж .; Насипури, Д. (1959). «Реакционные механизмы восстановления металл-аммиачными растворами». Тетраэдр. 6 (2): 148–153. Дои:10.1016/0040-4020(59)85008-0.
- ^ а б Бернем, Д. Р. (1969). «Ориентация в механизме березового восстановления анизола». Тетраэдр. 25 (4): 897–904. Дои:10.1016/0040-4020(69)85023-4.
- ^ Берч, А. Дж .; Субба Рао, Г. (1972). Adv. Орг. Chem. 8: 1–65. Отсутствует или пусто
| название =(Помогите) (и ссылки в нем) - ^ а б Берч, А. Дж .; Hinde, A. L .; Радом, Л. (1980). «Теоретический подход к восстановлению березы. Строение и устойчивость анион-радикалов замещенных бензолов». Варенье. Chem. Soc. 102 (10): 3370–3376. Дои:10.1021 / ja00530a012.
- ^ а б Берч, А. Дж .; Радом, Л. (1980). «Теоретический подход к восстановлению березы. Строение и стабильность циклогексадиенильных радикалов». Варенье. Chem. Soc. 102 (12): 4074–4080. Дои:10.1021 / ja00532a016.
- ^ а б Zimmerman, H.E .; Ван, П. А. (1990). «Региоселективность восстановления березы». Варенье. Chem. Soc. 112 (3): 1280–1281. Дои:10.1021 / ja00159a078.
- ^ а б Zimmerman, H.E .; Ван, П. А. (1993). «Региоселективность восстановления березы». Варенье. Chem. Soc. 115 (6): 2205–2216. Дои:10.1021 / ja00059a015.
- ^ Берч, А. Дж. (1992). «Стероидные гормоны и Люфтваффе. Предприятие по фундаментальным стратегическим исследованиям и некоторые из его последствий: сокращение Березовых превращается в сокращение рождаемости». Стероиды. 57 (8): 363–377. Дои:10.1016 / 0039-128X (92) 90080-S. PMID 1519267. S2CID 24827957. (показал механизм с мета)
- ^ Берч, А. Дж. (1996). «Березовая редукция в органическом синтезе». Pure Appl. Chem. 68 (3): 553–556. Дои:10.1351 / pac199668030553. S2CID 41494178. (все еще предлагает мета)
- ^ "Продвинутая органическая химия: реакции и синтез", Фрэнсис А. Кэри, Ричард Дж. Сандберг, стр. 437
- ^ Bachi, J. W .; Epstein, Y .; Herzberg-Minzly, H .; Левненталь, Дж. Э. (1969). «Синтез соединений, родственных гиббереллиновой кислоте. III. Аналоги кольца А гиббереллинов». J. Org. Chem. 34: 126–135. Дои:10.1021 / jo00838a030.
- ^ Табер, Д. Ф .; Gunn, B.P; Чинг Чиу, И. (1983). "Алкилирование аниона березовым восстановлением о-анисовой кислоты: 2-гептил-2-циклогексенон". Органический синтез. 61: 59.; Коллективный объем, 7, п. 249
- ^ Guo, Z .; Шульц, А. Г. (2001). «Методология органического синтеза. Получение и диастереоселективное восстановительное алкилирование березы 3-замещенных 2-метил-2,3-дигидроизоиндол-1-онов». J. Org. Chem. 66 (6): 2154–2157. Дои:10.1021 / jo005693g. PMID 11300915.
- ^ а б c Циммерман, Говард Э (1975). Квантовая механика для химиков-органиков. Нью-Йорк: Academic Press. стр.154–5. ISBN 0-12-781650-X.
- ^ а б c Циммерман, Х. Э. в «Молекулярных перестройках», Де Майо, П. Ред., Интерсайенс, Нью-Йорк, 1963, стр. 350–352.
- ^ Пауфлер, Р. М. (1960), доктор философии. Диссертация, Северо-Западный университет, Эванстон, Иллинойс.
- ^ Kuehne, M.E .; Ламберт, Б.Ф. (1963). «1,4-Дигидробензойная кислота». Органический синтез.; Коллективный объем, 5, п. 400
- ^ Paquette, L.A .; Барретт, Дж. Х. (1969). «2,7-Диметилоксепин». Органический синтез.; Коллективный объем, 5, п. 467
- ^ Табер, Д. Ф .; Gunn, B.P .; Чинг Чиу, И. (1983). "Алкилирование аниона березовым восстановлением о-анисовой кислоты: 2-гептил-2-циклогексенон". Органический синтез.; Коллективный объем, 7, п. 249
- ^ Клайв, Деррик Л. Дж. И Сунаси, Раджеш (2007). «Образование бензо-конденсированных карбоциклов формальной радикальной циклизацией на ароматическое кольцо». Органические буквы. 9 (14): 2677–2680. Дои:10.1021 / ol070849l. PMID 17559217.
- ^ Ecsery, Золтан и Мюллер, Миклош (1961). «Восстановление витамина D2 щелочными металлами». Мадьяр Кемай Фольойрат. 67: 330–332.
- ^ Донохо, Тимоти Дж. И Хаус, Дэвид (2002). «Безаммиачное частичное восстановление ароматических соединений с использованием ди-терт-бутилбифенил (LiDBB) ». Журнал органической химии. 67 (14): 5015–5018. Дои:10.1021 / jo0257593. PMID 12098328.
- ^ Гарст, Майкл Э .; Ллойд Дж .; Шервин; Н. Андрей; Натали С .; Альфред А .; и другие. (2000). «Уменьшение с помощью лития аминов с низкой молекулярной массой и этилендиамина». Журнал органической химии. 65 (21): 7098–7104. Дои:10.1021 / jo0008136. PMID 11031034.
- ^ Питерс, Байрон К .; Родригес, Кевин Х .; Reisberg, Solomon H .; Бейл, Себастьян Б .; Хики, Дэвид П .; Кавамата, Ю; Коллинз, Майкл; Старр, Джереми; Чен, Лонгруй; Удьявара, Сагар; Клундер, Кевин; Гори, Тимоти Дж .; Андерсон, Скотт Л .; Нейрок, Мэтью; Минтир, Шелли Д .; Баран, Фил С. (21 февраля 2019 г.). «Масштабируемое и безопасное синтетическое органическое электровосстановление, вдохновленное химией литий-ионных аккумуляторов». Наука. 363 (6429): 838–845. Дои:10.1126 / science.aav5606. ЧВК 7001862. PMID 30792297. Сложить резюме.
- ^ а б Берч, А. Дж. (1945). «Восстановление растворением металлов. Часть II». J. Chem. Soc.: 809. Дои:10.1039 / jr9450000809.
- ^ а б Берч, А. Дж. (1946). «Восстановление растворением металлов. Часть III». J. Chem. Soc.: 593. Дои:10.1039 / jr9460000593.
- ^ Берч, А. Дж. (1947). «Восстановление растворением металлов. Часть IV». J. Chem. Soc.: 102. Дои:10.1039 / jr9470000102.
- ^ Берч, Артур Дж. (1947). «Восстановление растворением металлов. Часть V». J. Chem. Soc.: 1642. Дои:10.1039 / jr9470001642.
- ^ Берч, А. Дж .; Мукерджи, С. М. (1949). «Восстановление растворением металлов. Часть VI. Некоторые применения в синтезе». J. Chem. Soc.: 2531. Дои:10.1039 / jr9490002531.
- ^ Wooster, C.B .; Годфри, К. Л. (1937). «Механизм восстановления ненасыщенных соединений щелочными металлами и водой». Журнал Американского химического общества. 59 (3): 596. Дои:10.1021 / ja01282a504.
- ^ Wilds, A. L .; Нельсон, Н.А. (1953). «Превосходный метод восстановления эфиров фенола до дигидропроизводных и ненасыщенных кетонов». Варенье. Chem. Soc. 75 (21): 5360–5365. Дои:10.1021 / ja01117a064.
- ^ Берч, А. Дж .; Смит, Х. (1958). «Восстановление металл-аминовыми растворами: приложения в синтезе и определении структуры». Кварта. Ред. (обзор). 12 (1): 17. Дои:10,1039 / qr9581200017.