Секвенирование третьего поколения - Third-generation sequencing
Секвенирование третьего поколения (также известный как долгосрочное секвенирование) является классом Секвенирование ДНК методы в настоящее время находятся в стадии активной разработки.[1]
Технологии секвенирования третьего поколения позволяют производить значительно более длинные считывания, чем секвенирование второго поколения.[1] Такое преимущество имеет решающее значение как для науки о геноме, так и для изучения биологии в целом. Однако данные секвенирования третьего поколения имеют гораздо более высокий уровень ошибок, чем предыдущие технологии, что может усложнить последующую сборку генома и анализ полученных данных.[2] Эти технологии активно развиваются, и ожидается, что они будут улучшены в отношении высокого уровня ошибок. Было обнаружено, что для приложений, которые более терпимы к частоте ошибок, таких как вызов структурных вариантов, секвенирование третьего поколения превосходит существующие методы.[нужна цитата ].
Современные технологии
Технологии секвенирования с подходом, отличным от платформ второго поколения, были впервые описаны как «третье поколение» в 2008–2009 годах.[3]
В настоящее время в центре разработки технологий секвенирования третьего поколения находятся несколько компаний, а именно: Тихоокеанские биологические науки, Оксфордская технология нанопор, Quantapore (Калифорния, США) и Stratos (Вашингтон, США). Эти компании используют принципиально разные подходы к секвенированию отдельных молекул ДНК.
PacBio разработала платформу секвенирования секвенирование одиночных молекул в реальном времени (SMRT), исходя из свойств нулевые волноводы. Сигналы представляют собой флуоресцентное излучение света от каждого нуклеотида, включенного ДНК-полимеразой, связанной со дном лунки zL.
Технология Oxford Nanopore’s включает прохождение молекулы ДНК через наноразмерную структуру пор с последующим измерением изменений электрического поля, окружающего пору; в то время как Quantapore использует другой собственный подход к нанопорам. Stratos Genomics разделяет основания ДНК полимерными вставками ",Xpandomers", чтобы обойти проблему" сигнал-шум "при чтении нанопор оцДНК.
Также примечателен Геликос подход к флуоресценции одной молекулы, но компания объявила о банкротстве в осень 2015 года.
Преимущества
Читает дольше
По сравнению с технологиями секвенирования текущего поколения, секвенирование третьего поколения имеет очевидное преимущество, заключающееся в получении гораздо более длинных чтений. Ожидается, что такая большая длина чтения облегчит многочисленные вычислительные проблемы, связанные с сборкой генома, реконструкцией транскриптов и метагеномикой среди других важных областей современной биологии и медицины.[1]
Хорошо известно, что геномы эукариот, включая приматов и человека, сложны и имеют большое количество длинных повторяющихся областей. Короткие чтения из секвенирования второго поколения должны прибегать к приближенным стратегиям, чтобы вывести последовательности на больших расстояниях для сборки и вызова генетических вариантов. Считывает конец пары были использованы секвенированием второго поколения для преодоления этих ограничений. Однако точные длины фрагментов на концах пары часто неизвестны и также должны быть приблизительно определены. Делая возможными длинные чтения, технологии секвенирования третьего поколения имеют явные преимущества.
Эпигенетика
Эпигенетические маркеры представляют собой стабильные и потенциально наследуемые модификации молекулы ДНК, не входящие в ее последовательность. Примером является метилирование ДНК в сайтах CpG, которое, как было установлено, влияет на экспрессию генов. Другой пример - модификации гистонов. Текущее поколение технологий секвенирования опирается на такие лабораторные методы, как ChIP-секвенирование для обнаружения эпигенетических маркеров. Эти методы включают маркировку нити ДНК, разрыв и фильтрацию фрагментов, содержащих маркеры, с последующим секвенированием. Секвенирование третьего поколения может позволить прямое обнаружение этих маркеров из-за их отличительного сигнала от других четырех нуклеотидных оснований.[4]
Портативность и скорость

Другие важные преимущества технологий секвенирования третьего поколения включают портативность и скорость секвенирования.[5] Поскольку требуется минимальная предварительная обработка образцов по сравнению с секвенированием второго поколения, может быть разработано меньшее оборудование. Компания Oxford Nanopore Technology недавно начала коммерциализацию Секвенсор MinION. Эта секвенсорная машина размером примерно с обычный USB-накопитель, и ее можно легко использовать, подключив к ноутбуку. Кроме того, поскольку процесс секвенирования не распараллеливается по участкам генома, данные можно собирать и анализировать в режиме реального времени. Эти преимущества секвенирования третьего поколения могут хорошо применяться в больничных условиях, где требуется быстрый сбор и анализ данных на месте.
Вызовы
Части этой статьи (те, которые относятся к технологиям секвенирования с длинным считыванием, обеспечивающим низкую точность считывания. Хотя 5 лет назад это было справедливо, циклическое согласованное считывание с секвенсором длительного считывания PacBio Sequel II может легко достичь даже более высокой точности считывания, чем сборка гибридного генома с комбинацией других секвенсоров. [1] PMID 31885515, 28364362, 31406327, 31897449, 31483244 ) нужно быть обновлено. (Январь 2020) |
Секвенирование третьего поколения в его нынешнем виде сталкивается с серьезными проблемами, в основном связанными с точной идентификацией нуклеотидных оснований; частота ошибок по-прежнему намного выше по сравнению с секвенированием второго поколения.[2] Обычно это происходит из-за нестабильности задействованного молекулярного механизма. Например, в технологии одиночного молекулярного секвенирования и секвенирования в реальном времени PacBio молекула ДНК-полимеразы становится все более поврежденной по мере того, как происходит процесс секвенирования.[2] Кроме того, поскольку процесс происходит быстро, сигналы отдельных баз могут быть размыты сигналами соседних баз. Это создает новую вычислительную задачу для расшифровки сигналов и, следовательно, определения последовательности. Такие методы как Скрытые марковские модели, например, с некоторым успехом использовались для этой цели.[4]
В среднем разные люди в человеческой популяции имеют около 99,9% общих генов. Другими словами, примерно только одна из тысячи баз будет отличаться у любых двух человек. Высокая частота ошибок, связанная с секвенированием третьего поколения, неизбежно создает проблемы для характеристики индивидуальных различий, существующих между членами одного и того же вида.
Сборка генома
Сборка генома реконструкция последовательностей ДНК всего генома. Обычно это делается с помощью двух принципиально разных подходов.
Справочное выравнивание
Когда доступен эталонный геном, как в случае человека, вновь секвенированные считывания можно просто сопоставить с эталонным геномом, чтобы охарактеризовать его свойства. Такая сборка на основе ссылок является быстрой и простой, но имеет недостаток, заключающийся в «сокрытии» новых последовательностей и вариантов с большим числом копий.Кроме того, для большинства организмов еще не существуют эталонные геномы.
De novo сборка
De novo Сборка - это альтернативный подход к сборке генома для эталонного выравнивания. Это относится к реконструкции последовательности всего генома полностью из считывания необработанной последовательности. Этот метод будет выбран, когда нет эталонного генома, когда вид данного организма неизвестен, как в метагеномика или когда существуют представляющие интерес генетические варианты, которые нельзя обнаружить при сравнительном выравнивании генома.
Учитывая короткие чтения, производимые современным поколением технологий секвенирования, сборка de novo представляет собой серьезную вычислительную проблему. Обычно к нему приближаются итеративный процесс поиска и соединения считываний последовательностей с разумными перекрытиями. Различные вычислительные и статистические методы, такие как графики де Брюйна и согласованные графики пересечения макетов были использованы для решения этой проблемы. Тем не менее, из-за очень повторяющейся природы эукариотических геномов точная и полная реконструкция геномных последовательностей при сборке de novo остается сложной задачей. Считывает конец пары были предложены как возможное решение, хотя точные длины фрагментов часто неизвестны и должны быть приблизительно определены.[6]
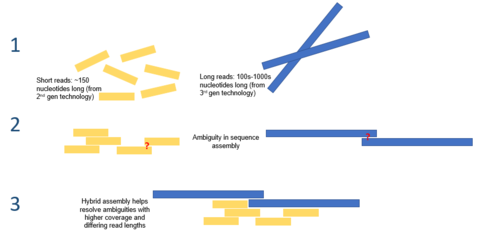
Гибридная сборка
Большая длина чтения, предлагаемая секвенированием третьего поколения, может облегчить многие из проблем, с которыми в настоящее время сталкиваются сборки генома de novo. Например, если целая повторяющаяся область может быть однозначно упорядочена за одно считывание, никаких вычислений не потребуется. Вычислительные методы были предложены, чтобы облегчить проблему высокой частоты ошибок. Например, в одном исследовании было продемонстрировано, что сборка микробного генома de novo с использованием одного только секвенирования PacBio работает лучше, чем секвенирование второго поколения.[7]
Секвенирование третьего поколения также может использоваться вместе с секвенированием второго поколения. Этот подход часто называют гибридным секвенированием. Например, длинные чтения из секвенирования третьего поколения могут использоваться для устранения неоднозначностей, которые существуют в геномах, ранее собранных с использованием секвенирования второго поколения. С другой стороны, короткие чтения второго поколения использовались для исправления ошибок, существующих в длинных чтениях третьего поколения. В целом было показано, что этот гибридный подход значительно улучшает сборки генома de novo.[8]
Эпигенетические маркеры
Метилирование ДНК (DNAm) - ковалентная модификация ДНК на сайтах CpG, что приводит к прикреплению метильные группы - это наиболее понятный компонент эпигенетический машины. Модификации ДНК и результирующая экспрессия генов могут варьироваться в зависимости от типа клеток, временного развития, с генетическим происхождением, могут изменяться из-за воздействия окружающей среды и передаются по наследству. После открытия ДНКм исследователи также обнаружили его связь с такими заболеваниями, как рак и аутизм.[9] В контексте этиологии этого заболевания DNAm является важным направлением дальнейших исследований.
Преимущества
Современные наиболее распространенные методы исследования состояния метилирования требуется анализ который фрагментирует ДНК перед стандартным секвенированием второго поколения на Иллюмина Платформа. В результате короткой длины чтения теряется информация о более длинных паттернах метилирования.[4] Технологии секвенирования третьего поколения предлагают возможность секвенирования одной молекулы в реальном времени для более длинных считываний и обнаружения модификации ДНК без вышеупомянутого анализа.[10]
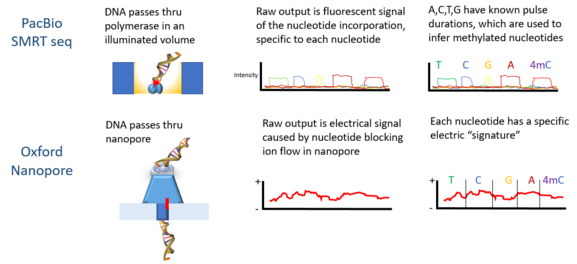
Oxford Nanopore Technologies » Миньон был использован для обнаружения DNAm. Когда каждая нить ДНК проходит через пору, она производит электрические сигналы, которые, как было установлено, чувствительны к эпигенетическим изменениям в нуклеотидах, и скрытая марковская модель (HMM) использовался для анализа данных MinION для обнаружения 5-метилцитозин (5mC) Модификация ДНК.[4] Модель обучалась с использованием синтетически метилированных Кишечная палочка ДНК и полученные сигналы измеряются с помощью нанопор. Затем обученную модель использовали для обнаружения 5mC в геномных считываниях MinION из линии клеток человека, которая уже имела эталонный метилом. Классификатор имеет точность 82% на случайно выбранных одноэлементных сайтах, которая увеличивается до 95% при применении более строгих пороговых значений.[4]
Другие методы предназначены для различных типов модификаций ДНК с использованием платформы MinION. Stoiber et al. исследовали 4-метилцитозин (4mC) и 6-метиладенин (6mA) вместе с 5mC, а также создали программное обеспечение для прямой визуализации необработанных данных MinION удобным для человека способом.[11] Здесь они обнаружили, что в Кишечная палочка, имеющий известную метилом, окна событий длиной 5 пар оснований могут использоваться для разделения и статистического анализа необработанных электрических сигналов MinION. Простой U-критерий Манна-Уитни может обнаруживать измененные части Кишечная палочка последовательности, а также дальнейшее разделение модификаций на области 4mC, 6mA или 5mC.[11]
Вполне вероятно, что в будущем необработанные данные MinION будут использоваться для обнаружения множества различных эпигенетических меток в ДНК.
PacBio секвенирование также использовалось для обнаружения метилирования ДНК. В этой платформе ширина импульса - ширина импульса флуоресцентного света - соответствует определенной базе. В 2010 году было показано, что расстояние между импульсами в контрольных и метилированных образцах различается, и для каждого типа метилирования существует «сигнатурная» длительность импульса.[10] В 2012 году с помощью платформы PacBio сайты связывания ДНК метилтрансферазы были охарактеризованы.[12] Обнаружение N6-метилирования в C Elegans был показан в 2015 году.[13] Метилирование ДНК на N6-аденин с использованием платформы PacBio в мыши эмбриональные стволовые клетки был показан в 2016 году.[14]
Другие формы модификаций ДНК - от тяжелых металлов, окисления или УФ-повреждения - также являются возможными направлениями исследований с использованием секвенирования третьего поколения Oxford Nanopore и PacBio.
Недостатки
Обработка необработанных данных - например, нормализация к среднему сигналу - была необходима для необработанных данных MinION, что ограничивало возможности технологии в реальном времени.[11] Согласованность электрических сигналов по-прежнему остается проблемой, что затрудняет точное определение нуклеотида. MinION имеет низкую пропускную способность; поскольку трудно получить множественные перекрывающиеся считывания, это дополнительно приводит к проблемам с точностью обнаружения последующих модификаций ДНК. Как скрытая марковская модель, так и статистические методы, используемые с необработанными данными MinION, требуют повторных наблюдений модификаций ДНК для обнаружения, а это означает, что отдельные модифицированные нуклеотиды должны постоянно присутствовать в нескольких копиях генома, например в нескольких клетках или плазмидах в образце.
Для платформы PacBio, в зависимости от того, какое метилирование вы ожидаете обнаружить, потребности в покрытии могут различаться. По состоянию на март 2017 года другие эпигенетические факторы, такие как модификации гистонов, не были обнаружены с помощью технологий третьего поколения. Более длинные паттерны метилирования часто теряются, потому что меньшие контиги все еще нуждаются в сборке.
Транскриптомика
Транскриптомика это исследование транскриптом, обычно путем характеристики относительного содержания молекул матричной РНК в исследуемой ткани. Согласно центральная догма молекулярной биологии генетическая информация передается от молекул двухцепочечной ДНК к одноцепочечным молекулам мРНК, где они могут быть легко переведены в функциональные белковые молекулы. Изучая транскриптом, можно получить ценную информацию о регуляции экспрессии генов.
Хотя уровни экспрессии как уровень гена можно более или менее точно отобразить с помощью секвенирования второго поколения, информация об уровне транскрипта все еще остается важной проблемой.[15] Как следствие, роль альтернативного сплайсинга в молекулярной биологии остается в значительной степени неуловимой. Технологии секвенирования третьего поколения открывают многообещающие перспективы в решении этой проблемы, позволяя секвенировать молекулы мРНК по всей их длине.
Альтернативная сварка
Альтернативная сварка (AS) - это процесс, с помощью которого один ген может дать начало множеству различных транскриптов мРНК и, следовательно, различным трансляциям белков.[16] Некоторые данные свидетельствуют о том, что AS является повсеместным явлением и может играть ключевую роль в определении фенотипов организмов, особенно у сложных эукариот; все эукариоты содержат гены, состоящие из интронов, которые могут подвергаться AS. В частности, было подсчитано, что AS встречается в 95% всех мультиэкзонных генов человека.[17] AS имеет неоспоримый потенциал влиять на множество биологических процессов. Расширение знаний в этой области имеет решающее значение для изучения биологии в целом.
Реконструкция стенограммы
Текущее поколение технологий секвенирования производит только короткие чтения, что накладывает огромные ограничения на способность обнаруживать отдельные транскрипты; короткие чтения должны быть преобразованы в исходные расшифровки, которые могли бы дать повод для результирующих наблюдений чтения.[18] Эта задача еще более усложняется из-за очень вариабельных уровней экспрессии в транскриптах и, как следствие, вариабельных покрытий чтения в последовательности гена.[18] Кроме того, экзоны могут быть общими для отдельных транскриптов, что делает однозначные выводы практически невозможными.[16] Существующие вычислительные методы делают выводы, основанные на накоплении коротких считываний в различных местах последовательности, часто с помощью упрощающих предположений.[18] Запонки использует экономный подход, стремясь объяснить все чтения с наименьшим возможным количеством расшифровок.[19] С другой стороны, StringTie пытается одновременно оценить количество транскриптов во время сборки считываний.[18] Эти методы, хотя и разумны, не всегда могут идентифицировать настоящие стенограммы.
В исследовании, опубликованном в 2008 году, было изучено 25 различных существующих протоколов реконструкции стенограммы.[15] Его данные свидетельствуют о том, что существующие методы, как правило, неэффективны при сборке транскриптов, хотя способность обнаруживать отдельные экзоны относительно не нарушена.[15] По оценкам, средняя чувствительность к обнаружению экзонов по 25 протоколам составляет 80% для Caenorhabditis elegans гены.[15] Для сравнения, чувствительность идентификации транскриптов снижается до 65%. Для человека в исследовании сообщается, что чувствительность обнаружения экзонов в среднем составляет 69%, а чувствительность обнаружения транскриптов составляет в среднем всего 33%.[15] Другими словами, для человека существующие методы способны идентифицировать менее половины всех существующих расшифровок.
Технологии секвенирования третьего поколения продемонстрировали многообещающие перспективы в решении проблемы обнаружения транскриптов, а также оценки количества мРНК на уровне транскриптов. Несмотря на то, что частота ошибок остается высокой, технологии секвенирования третьего поколения могут обеспечивать гораздо более длинные считывания.[20] Pacific Bioscience представила платформу iso-seq, предлагая секвенировать молекулы мРНК по всей их длине.[20] Ожидается, что Oxford Nanopore предложит аналогичные технологии. Проблема с более высоким коэффициентом ошибок может быть уменьшена за счет дополнительных высококачественных коротких чтений. Этот подход был ранее протестирован, и было сообщено, что он снижает частоту ошибок более чем в 3 раза.[21]
Метагеномика
Метагеномика это анализ генетического материала, полученного непосредственно из образцов окружающей среды.
Преимущества
Главное преимущество технологий секвенирования третьего поколения в метагеномика это их скорость секвенирования по сравнению с методами второго поколения. Скорость секвенирования важна, например, в клинических условиях (т.е. возбудитель идентификация), чтобы обеспечить эффективную диагностику и своевременные клинические действия.
MinION от Oxford Nanopore использовался в 2015 году для метагеномного обнаружения патогенов в режиме реального времени в сложных клинических образцах с высоким уровнем фона. Первый Вирус Эбола (EBV) чтение было секвенировано через 44 секунды после сбора данных.[22] Было единообразное отображение чтения в геном; по крайней мере, одно чтение соответствует> 88% генома. Относительно длинные считывания позволили секвенировать почти полный вирусный геном с высокой точностью (97–99% идентичности) непосредственно из первичного клинического образца.[22]
Обычный филогенетический Маркер для исследований разнообразия микробного сообщества - это 16S рибосомная РНК ген. Платформа SMRT MinION и PacBio была использована для секвенирования этого гена.[23][24] В этом контексте частота ошибок PacBio была сопоставима с частотой более коротких чтений из 454 и платформы секвенирования MiSeq компании Illumina.[нужна цитата ]
Недостатки
Высокая частота ошибок MinION (~ 10-40%) не позволила идентифицировать устойчивость к противомикробным препаратам маркеры, для которых необходимо разрешение отдельных нуклеотидов. По той же причине, эукариотический возбудители не выявлены.[22] Легкость переноса загрязнений при повторном использовании той же проточной кюветы (стандартные протоколы промывки не работают) также вызывает беспокойство. Уникальные штрих-коды могут позволить большее мультиплексирование. Кроме того, выполнение точной идентификации видов для бактерии, грибы и паразиты очень сложно, поскольку они разделяют большую часть генома, а некоторые отличаются только на <5%.
Стоимость базового секвенирования по-прежнему значительно выше, чем у MiSeq. Однако перспектива дополнения справочных баз данных полноразмерными последовательностями организмов ниже предела обнаружения из Sanger подход;[23] это могло бы значительно помочь в идентификации организмов в метагеномике.
Рекомендации
- ^ а б c Блейдорн, Кристоф (2016-01-02). «Третье поколение секвенирования: технология и ее потенциальное влияние на исследования эволюционного биоразнообразия». Систематика и биоразнообразие. 14 (1): 1–8. Дои:10.1080/14772000.2015.1099575. ISSN 1477-2000.
- ^ а б c Гупта, Пушпендра К. (01.11.2008). «Технологии секвенирования одномолекулярной ДНК для будущих исследований в области геномики». Тенденции в биотехнологии. 26 (11): 602–611. Дои:10.1016 / j.tibtech.2008.07.003. PMID 18722683.
- ^ Отметьте Хайден, Эрика (2009-02-06). «Секвенирование генома: третье поколение». Новости природы. 457 (7231): 768–769. Дои:10.1038 / новости.2009.86. PMID 19212365.
- ^ а б c d е Симпсон, Джаред Т .; Уоркман, Рэйчел; Zuzarte, Philip C .; Давид, Матей; Дурси, Льюис Джонатан; Тимп, Уинстон (2016-04-04). «Обнаружение метилирования ДНК с использованием секвенатора Oxford Nanopore Technologies MinION». bioRxiv 10.1101/047142.
- ^ Schadt, E. E .; Тернер, С .; Касарскис, А. (15.10.2010). «Окно в секвенирование третьего поколения». Молекулярная генетика человека. 19 (R2): R227 – R240. Дои:10,1093 / hmg / ddq416. ISSN 0964-6906. PMID 20858600.
- ^ Ли, Жуйцян; Чжу, Хунмэй; Руан, Цзюэ; Цянь, Вубин; Фанг, Сяодун; Ши, Чжунбинь; Ли, Инжруй; Ли, Шэнтин; Шан, Гао (01.02.2010). «Сборка de novo человеческих геномов с массовым параллельным секвенированием короткого чтения». Геномные исследования. 20 (2): 265–272. Дои:10.1101 / гр.097261.109. ISSN 1088-9051. ЧВК 2813482. PMID 20019144.
- ^ Чин, Чен-Шань; Александр, Дэвид Х .; Маркс, Патрик; Кламмер, Аарон А .; Дрейк, Джеймс; Хайнер, Шерил; Клам, Алисия; Коупленд, Алекс; Хаддлстон, Джон (01.06.2013). «Негибридные, готовые сборки микробного генома на основе данных секвенирования SMRT». Природные методы. 10 (6): 563–569. Дои:10.1038 / nmeth.2474. ISSN 1548-7091. PMID 23644548.
- ^ Гудвин, Сара; Гуртовски, Джеймс; Эте-Сэйерс, Скотт; Дешпанде, Панчаджанья; Schatz, Майкл С .; Маккомби, У. Ричард (01.11.2015). «Секвенирование Oxford Nanopore, исправление гибридных ошибок и сборка de novo эукариотического генома». Геномные исследования. 25 (11): 1750–1756. Дои:10.1101 / гр.191395.115. ISSN 1088-9051. ЧВК 4617970. PMID 26447147.
- ^ Фрейзер, Хантер Б .; Lam, Lucia L .; Neumann, Сара М .; Кобор, Майкл С. (09.02.2012). «Популяционная специфичность метилирования ДНК человека». Геномная биология. 13 (2): R8. Дои:10.1186 / gb-2012-13-2-r8. ISSN 1474-760X. ЧВК 3334571. PMID 22322129.
- ^ а б Flusberg, Benjamin A .; Webster, Dale R .; Ли, Джессика Х .; Трэверс, Кевин Дж .; Olivares, Eric C .; Кларк, Тайсон А .; Корлах, Йонас; Тернер, Стивен В. (01.06.2010). «Прямое обнаружение метилирования ДНК во время секвенирования одной молекулы в реальном времени». Природные методы. 7 (6): 461–465. Дои:10.1038 / nmeth.1459. ЧВК 2879396. PMID 20453866.
- ^ а б c Stoiber, Marcus H .; Быстрее, Джошуа; Иган, Роб; Ли, Джи Ын; Celniker, Susan E .; Нили, Роберт; Ломан, Николай; Пеннаккио, Лен; Браун, Джеймс Б. (15 декабря 2016 г.). «De novo идентификация модификаций ДНК, обеспечиваемых геномной обработкой сигнала нанопор». bioRxiv 10.1101/094672.
- ^ Clark, T. A .; Мюррей, И. А .; Morgan, R.D .; Кислюк, А.О .; Spittle, K. E .; Boitano, M .; Фоменков, А .; Робертс, Р. Дж .; Корлач, Дж. (01.02.2012). «Определение специфичности ДНК-метилтрансферазы с помощью одномолекулярного секвенирования ДНК в реальном времени». Исследования нуклеиновых кислот. 40 (4): e29. Дои:10.1093 / нар / gkr1146. ISSN 0305-1048. ЧВК 3287169. PMID 22156058.
- ^ Грир, Эрик Либерман; Бланко, Марио Андрес; Гу, Лей; Сендинк, Эрдем; Лю, Цзяньчжао; Аристизабаль-Корралес, Давид; Сюй, Чи-Хунг; Aravind, L .; Он, Чуан (2015). «Метилирование ДНК по N6-аденину у C. elegans». Клетка. 161 (4): 868–878. Дои:10.1016 / j.cell.2015.04.005. ЧВК 4427530. PMID 25936839.
- ^ Wu, Tao P .; Ван, Дао; Seetin, Matthew G .; Лай, Юнцюань; Чжу, Шицзя; Линь, Кайсюань; Лю, Ифэй; Byrum, Stephanie D .; Макинтош, Сэмюэл Г. (21 апреля 2016 г.). «Метилирование ДНК по N6-аденину в эмбриональных стволовых клетках млекопитающих». Природа. 532 (7599): 329–333. Bibcode:2016 Натур.532..329Вт. Дои:10.1038 / природа17640. ISSN 0028-0836. ЧВК 4977844. PMID 27027282.
- ^ а б c d е Стейджер, Тамара; Abril, Josep F .; Engström, Pär G .; Кокоцински, Феликс; Консорциум RGASP; Хаббард, Тим Дж .; Гиго, Родерик; Харроу, Дженнифер; Бертоне, Пол (2013-12-01). «Оценка методов реконструкции транскриптов для RNA-seq». Природные методы. 10 (12): 1177–1184. Дои:10.1038 / nmeth.2714. ISSN 1548-7091. ЧВК 3851240. PMID 24185837.
- ^ а б Грейвли, Брентон Р. (2001). «Альтернативный сплайсинг: увеличение разнообразия в протеомном мире». Тенденции в генетике. 17 (2): 100–107. Дои:10.1016 / s0168-9525 (00) 02176-4. PMID 11173120.
- ^ Пан, Цюнь; Шай, Офер; Ли, Лео Дж .; Фрей, Брендан Дж .; Бленкоу, Бенджамин Дж. (01.12.2008). «Глубокое исследование альтернативной сложности сплайсинга в человеческом транскриптоме с помощью высокопроизводительного секвенирования». Природа Генетика. 40 (12): 1413–1415. Дои:10,1038 / нг.259. ISSN 1061-4036. PMID 18978789.
- ^ а б c d Пертя, Михаэла; Pertea, Geo M .; Антонеску, Корина М .; Чанг, Цзун-Ченг; Mendell, Joshua T .; Зальцберг, Стивен Л. (2015-03-01). «StringTie обеспечивает улучшенную реконструкцию транскриптома из считываний RNA-seq». Природа Биотехнологии. 33 (3): 290–295. Дои:10.1038 / nbt.3122. ISSN 1087-0156. ЧВК 4643835. PMID 25690850.
- ^ Трапнелл, Коул; Уильямс, Брайан А .; Pertea, Geo; Мортазави, Али; Кван, Гордон; van Baren, Marijke J .; Зальцберг, Стивен Л .; Уолд, Барбара Дж .; Пахтер, Лиор (01.05.2010). «Сборка и количественная оценка транскриптов с помощью RNA-Seq выявляет неаннотированные транскрипты и переключение изоформ во время дифференцировки клеток». Природа Биотехнологии. 28 (5): 511–515. Дои:10.1038 / nbt.1621. ISSN 1087-0156. ЧВК 3146043. PMID 20436464.
- ^ а б Abdel-Ghany, Salah E .; Гамильтон, Майкл; Якоби, Дженнифер Л .; Нгам, Питер; Девитт, Николас; Шилки, Фэй; Бен-Гур, Аса; Редди, Аниредди С. Н. (24.06.2016). «Обзор транскриптома сорго с использованием длинных считываний одиночных молекул». Nature Communications. 7: 11706. Bibcode:2016НатКо ... 711706A. Дои:10.1038 / ncomms11706. ISSN 2041-1723. ЧВК 4931028. PMID 27339290.
- ^ Ау, Кин Фай; Андервуд, Джейсон Дж .; Ли, Лоуренс; Вонг, Вин Хунг (2012-10-04). «Повышение точности длительного считывания PacBio за счет выравнивания короткого считывания». PLOS ONE. 7 (10): e46679. Bibcode:2012PLoSO ... 746679A. Дои:10.1371 / journal.pone.0046679. ISSN 1932-6203. ЧВК 3464235. PMID 23056399.
- ^ а б c Greninger, Александр Л .; Naccache, Samia N .; Федерман, Скотт; Ю, Гиксиа; Mbala, Placide; Брес, Ванесса; Страйк, Дуг; Букет, Джером; Сомасекар, Снеха (01.01.2015). «Быстрая метагеномная идентификация вирусных патогенов в клинических образцах с помощью анализа секвенирования нанопор в реальном времени». Геномная медицина. 7: 99. Дои:10.1186 / s13073-015-0220-9. ISSN 1756-994X. ЧВК 4587849. PMID 26416663.
- ^ а б Schloss, Patrick D .; Jenior, Matthew L .; Koumpouras, Charles C .; Весткотт, Сара Л .; Горец, Сара К. (01.01.2016). «Секвенирование фрагментов гена 16S рРНК с использованием системы секвенирования ДНК PacBio SMRT». PeerJ. 4: e1869. Дои:10.7717 / peerj.1869. ЧВК 4824876. PMID 27069806.
- ^ Бенитес-Паес, Альфонсо; Portune, Кевин Дж .; Санс, Иоланда (01.01.2016). «Разрешение на уровне видов ампликонов гена 16S рРНК, секвенированных с помощью портативного секвенатора нанопор MinION ™». GigaScience. 5: 4. Дои:10.1186 / s13742-016-0111-z. ISSN 2047-217X. ЧВК 4730766. PMID 26823973.