Дивинилциклопропан-циклогептадиеновая перегруппировка - Divinylcyclopropane-cycloheptadiene rearrangement
В дивинилциклопропан-циклогептадиеновая перегруппировка представляет собой органическое химическое превращение, которое включает изомеризацию 1,2-дивинилциклопропана в циклогептадиен или -триен. Это концептуально связано с Коуп перестановка, но имеет преимущество сильной термодинамической движущей силы из-за снятия кольцевой деформации. Эта термодинамическая мощность в последнее время рассматривается как альтернативный источник энергии.[1]
Вступление
В 1960 году Фогель обнаружил, что 1,2-дивинилциклопропан перегруппировывается в циклогептан-1,4-диен.[2] После его открытия в 1960-х годах последовала серия интенсивных механистических исследований реакции, поскольку исследователи поняли, что она имеет сходство (как структурное, так и механистическое) с соответствующими реакциями. перестановка винилциклопропана в циклопентен. К 1970-м годам перегруппировка достигла синтетической полезности.[3] и по сей день он продолжает оставаться полезным методом образования семичленных колец. Сообщалось о вариантах, включающих гетероатомы (см. Ниже).
(1)

Преимущества: Будучи перегруппировкой, процесс демонстрирует идеальную атомную экономию. Часто это происходит спонтанно, без использования катализатора. Конкуренция для полностью углеродной перегруппировки минимальна.
Недостатки: Во многих случаях конфигурацию исходных материалов необходимо контролировать -транс-дивинилциклопропаны часто требуют нагревания для облегчения изомеризации до того, как произойдет перегруппировка. Перегруппировки с участием гетероатомов могут приводить к снижению выходов из-за образования побочных продуктов.
Механизм и стереохимия
Преобладающий механизм
Основная дискуссия о механизме перегруппировки сосредоточена на том, согласованный (сигматропный) или ступенчатый (бирадикальный) процесс. Механистические эксперименты показали, что транс-дивинилциклопропаны эпимеризуются до соответствующих СНГ изомеры и претерпевают перегруппировку по наиболее вероятному согласованному пути.[4][5] Было предложено лодкообразное переходное состояние, которое помогает объяснить наблюдаемую стереоспецифичность процесса. Была ли начальная эпимеризация транс субстрат происходит посредством одно- или двухцентрового процесса, в большинстве случаев неясно.
(2)

Варианты перегруппировки, катализируемые переходными металлами, известны, и механизмы варьируются. В одном примере с использованием бис (этилен) гексафторацетилацетоната родия координация и образование бис-π-аллильного комплекса предшествуют электроциклическому замыканию кольца и высвобождению катализатора.[6]
(3)

Стереоселективные варианты
Реакции дивинилциклопропанов, содержащих замещенные двойные связи, стереоспецифичны по отношению к конфигурациям двойных связей -СНГ,СНГ изомеры дают СНГ продукты, а СНГ,транс изомеры дают транс-продукты. Таким образом, хиральные нерацемические исходные материалы дают хиральные продукты без потери энантиомерной чистоты. В приведенном ниже примере в каждом случае наблюдались только изображенные изомеры.[7]
(4)

Объем и ограничения
В названную реакцию вступает большое количество дивинилциклопропанов. Эти предшественники были получены различными методами, включая добавление циклопропилнуклеофилов (солей лития,[8] или медь[9]) к активированным двойным или тройным связям, отщепление бис (2-галогенэтил) циклопропанов[10] и циклопропанирование.[11]
В приведенном ниже примере при добавлении-отщеплении купрата образуется временный енон. 1, который перестраивается в спироцикл 2.
(5)
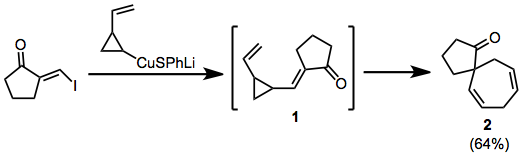
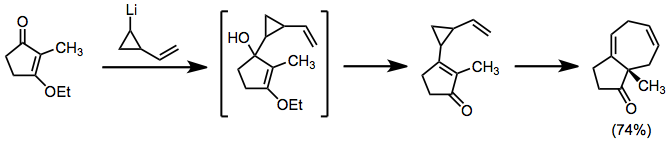
Литийорганические соединения могут использоваться в аналогичной роли, но непосредственно добавляются к карбонилам. Результат продуктов с объединенной топологией.[8]
(6)
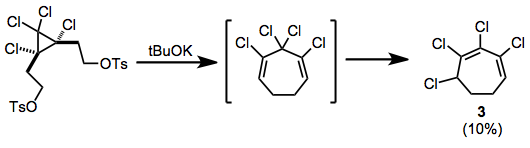
Наблюдалась перегруппировка после удаления дитозилатов; полученный таким образом хлорированный циклогептадиен изомеризуется в сопряженный гептадиен. 3 во время реакции.[10]
(7)
Циклопропанирование конъюгированными диазосоединениями дает дивинилциклопропаны, которые затем подвергаются перегруппировке. При использовании циклических исходных материалов образуются мостиковые продукты.[12]
(8)

Субстраты, содержащие трехчленные гетероциклические кольца, также могут подвергаться реакции. СНГ-Дивинилэпоксиды дают оксепины при повышенных температурах (100 ° C). транс Изомеры претерпевают интересную конкурентную перегруппировку в дигидрофураны через посредство карбонильной группы. илида[13] и тот же промежуточный илид был предложен в качестве прямого предшественника оксепинового продукта 4.[14] Конъюгированные диенилэпоксиды образуют аналогичные продукты, что подтверждает существование промежуточного илидного соединения.[15]
(9)
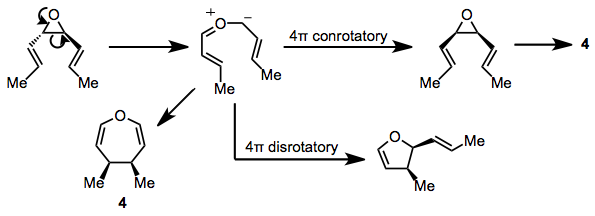
Дивинил азиридины проходят аналогичный набор реакций, обеспечивая азепины или винил пирролины в зависимости от относительной конфигурации исходного материала азиридина.[16] Дивинил тиираны может обеспечить тиепины или дигидротиофены, хотя эти реакции протекают медленнее, чем реакции соответствующих азот- и кислородсодержащих соединений.
Синтетические приложения
Самое раннее наблюдение циклогептадиена через перегруппировку названия было сделано Байером в его синтезе Eucarvone из гидробромида карвона.[17] Механистические исследования показали, что перестройка действительно происходила через согласованный механизм типа Копа.[18]
(10)

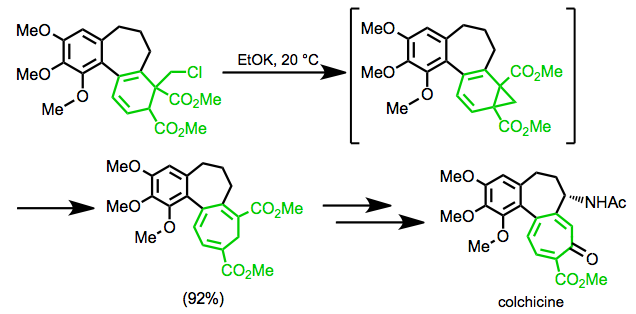
В синтезе Эшенмозера колхицин, перегруппировка используется для образования семичленного кольца мишени.[19]
(11)
В рацемическом синтезе сиренина используется Реакция Виттига с образованием ключевого дивинилциклопропана. Гидрирование продукта перегруппировки дало мишень.[20]
(12)

Условия и методика экспериментов
Типичные условия
Обычно перегруппировку проводят сразу после образования дивинилциклопропана в том же сосуде. Иногда необходимо отопление, особенно для транс субстраты, которые должны подвергнуться эпимеризации перед перегруппировкой. Однако при наличии энергии, достаточной для преодоления активационных барьеров, изомеризация обычно очень эффективна.
Пример процедуры[21]
(13)

К холодному (–78 °) перемешанному раствору диизопропиламид лития (1,4–1,5 ммоль / ммоль кетона) в сухом THF (4 мл / ммоль основания) в атмосфере аргона медленно добавляли раствор н-бутил-транс-2-винилциклопропилкетона (1,19 ммоль) в сухом растворе. THF (1 мл / ммоль кетона), и полученный раствор перемешивали при –78 ° в течение 45 минут. Раствор свежесублимированного трет-бутилдиметилсилилхлорида (1,6 ммоль / ммоль кетона) в сухом THF (1 мл / ммоль хлорида) с последующим сухим HMPA (0,5 мл / ммоль кетона). Раствор перемешивали при -78 ° в течение 15 минут и при комнатной температуре в течение 2-3 часов, а затем распределяли между насыщенными водными растворами. бикарбонат натрия и пентан (10 мл и 20 мл / ммоль кетона соответственно). Водную фазу дважды промывали пентаном. Объединенный экстракт промывали четыре раза насыщенным водным раствором бикарбоната натрия и дважды промывали рассол, а затем сушить (MgSO4 ). Удаление растворителя с последующей перегонкой оставшегося масла из колбы в колбу дало соответствующее силиловый эфир енола в виде бесцветного масла, которое не проявляет поглощения растяжения карбонила в ИК-диапазоне. Термолиз простого эфира силил енола осуществляли нагреванием (чистый, в атмосфере аргона) при 230 ° (температура воздушной бани) в течение 30-60 минут. Прямая перегонка (140–150 ° / 12 торр) полученных материалов дала циклогептадиен с выходом 85%: ИК (пленка) 1660, 1260, 840 см – 1; 1H ЯМР (CDCl3 ) δ 0,09 (с, 6H), 0,88 (с, 9H), 0,7–2,75 (м, 14H), 4,8 (т, 1H, J = 5,5 Гц), 5,5–5,9 (м, 2H).
Рекомендации
- ^ Худлики, Т .; Fan, R .; Reed, J. W .; Гадамасетти, К.Г. Орг. Реагировать. 1992, 41, 1-133. Дои:10.1002 / 0471264180.or041.01
- ^ Фогель, Э. Энгью. Chem. 1960, 72, 4.
- ^ Wender, P.A .; Eissenstat, M. A .; Филоса, М. П. Варенье. Chem. Soc. 1979, 101, 2196.
- ^ Arai, M .; Кроуфорд, Р. Дж. Может. J. Chem. 1972, 50, 2158.
- ^ Болдуин, Дж. Э .; Флеминг, Р. Х.Варенье. Chem. Soc. 1973, 95, 5256.
- ^ Alcock, N.W .; Brown, J.M .; Conneely, J. A .; Стофко младший, Дж. Дж. J. Chem. Soc., Chem. Commun., 1975, 234.
- ^ Brule, D .; Chalchat, J.C .; Весьер, Р. Бык. Soc. Чим. Пт. 1978, Нет. 7-8, II-385.
- ^ а б Wender, P.A .; Филоса, М. П. J. Org. Chem. 1976, 41, 3490.
- ^ Marino, J.P .; Браун, Л. Дж. Tetrahedron Lett. 1976, 3245.
- ^ а б Muller, P .; Рей, М. Helv. Чим. Acta, 1982, 65, 1191.
- ^ Худлики, Т .; Рулин, Ф .; Лавлейс, Т .; Рид, Дж. У. в Исследования в области химии природных продуктов, Атта-ур-Рахман, изд., Elsevier, Амстердам, 1989 г., часть B, стр. 3.
- ^ Дэвис, Х. М. Л .; Кларк, Д. М .; Смит, Т.К. Tetrahedron Lett. 1985, 26, 5659.
- ^ Pommelet, J.C .; Manisse, N .; Чучи, Дж. Тетраэдр, 1972, 28, 3929.
- ^ Браун, Р.А. J. Org. Chem. 1963, 28, 1383.
- ^ Eberbach, W .; Розер, Дж. Tetrahedron Lett. 1987, 28, 2685.
- ^ Manisse, N .; Чучи, Дж. Тетраэдр, 1977, 33, 2399.
- ^ Байер, А. Бер. 1894, 27, 810; там же. 1898, 31, 2067.
- ^ Vogel, E .; Отт, К.-Х .; Гайек, К. Justus Liebigs Ann. Chem. 1961, 644, 172.
- ^ Schreiber, von J .; Leimgruber, W .; Pesaro, M .; Schudel, P .; Threlfall, T .; Эшенмозер, А. Helv. Чим. Acta 1961, 44, 540.
- ^ Jaenicke, L .; Акинтоби, Т .; Мюллер, Д. Энгью. Chem. Int. Эд. Англ. 1971, 10, 492.
- ^ Пирс, Э .; Burmeister, M. S .; Рейссиг, Х.У. Может. J. Chem. 1986, 64, 180.